重铬酸钾容量分析用标准溶液干啥用的呢?
滴定分析容量分析用标准溶液的制备谢谢
[size=4] 在日常化学分析中用[B]容量法[/B]进行[B]滴定[/B]时,标准滴定溶液是用“[B]滴定度[/B]”表示还是用“[B]物质的量浓度[/B]”形式表示? 哪种形式表示更便于分析结果的计算?[/size]
因为省事我们习惯性在配好标准溶液后就直接用容量瓶存放。近日有人建议不要用容量瓶存放标准溶液。请问你们是用什么存放标准溶液(特别是痕量分析所用标液)。用这些器皿有什么好处与坏处?谢谢!
在定量分析中, 大家都用什么溶剂来配标准溶液. 有人用MeOH, 有人用DMSO, 有人用DMF.每个人都有一套说法. 说一说你都用什么溶剂配标准溶液?
标准溶液要求朔源,一般用纯金属或者基准试剂配制,今天看见有人用分析纯试剂配制标准溶液,很是恼火。既然人家这样配制标准溶液,肯定有一定的原因,但是我很是不解,欢迎大家讨论,分析纯试剂是否可以配制标准溶液?
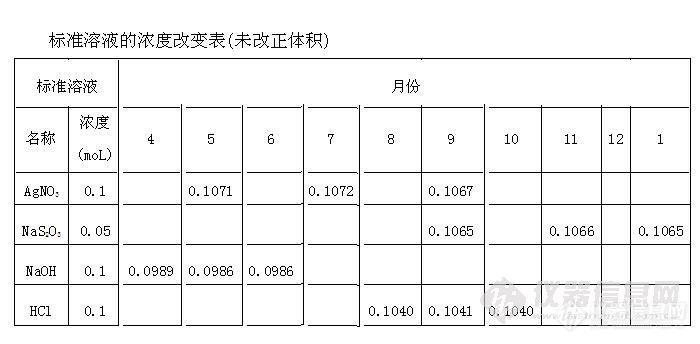
在容量分析中需注意的问题用一种已知浓度的标准溶液滴加到被测物的溶液中,以指示剂来判断滴定终点,由标准溶液的用量及浓度计算出被测物含量的方法,叫容量分析法。一般将容量分析法分为中和法、氧化还原法、沉淀法及络合滴定法等。一、误差在容量分析中,体积的测量和终点的确定等都可能发生误差,主要来源有下列几种:(一)、仪器误差:使用未校准过的仪器测量体积所产生的误差,称为仪器误差。(二)、读数误差:读取溶液体积时所产生的误差,称为读数误差。读数误差的相对值随着溶液用量的增加而减少。滴定读数允许误差的绝对值为0.02毫升,因此,标准溶液体积达到20毫升时(0.02/20×100=0.1%),方能符合容量分析的要求。(三)、滴定误差:由于滴定终点不可能恰好在等当点上,亦即指示剂颜色的改变不是恰好与等当点完全符合,因此可能产生滴定误差。为了减少容量分析中的误差,除容量仪器须经过校准外,还应注意选择合适的指示剂。所用标准溶液的浓度,随放置时间可能有改变,应定期校准。滴定时应特别注意下列几点问题;1、每次滴定最好从滴定管刻度的零点开始,因为这样每次滴定时的读数都在滴定管刻度的同一部分,由刻度不准确而引起的误差就可以抵消。2、滴定时应控制溶液滴出的速度不要大于每秒3~4滴,否则就会有少许溶液留在管壁上,使读数不准确。如滴定进行较快,应当等候1~2分钟,使管壁的溶液充分流下来后再读数。滴定接近终点时,滴定速度尤其要放慢,可控制溶液半滴半滴地滴入,以便能准确地观察终点的变化。3、为了减少滴定误差,滴定时用去溶液的体积应为滴定管容量的1/2或2/3。溶液用量超过滴定管容量,将增大误差;但如溶液用量太少,则读数的百分误差也将增大。滴定管应与用去滴定液的体积协调一致。即滴定液5毫升(或10毫升)时,只能用5毫升或10毫升的滴定管,滴定液超过10毫升时,可用25毫升或50毫升滴定管,这样可避免产生较大的读数误差。例如:在滴定合成水样的总硬度时,仅需滴定少于5毫升的EDTA标准溶液,但使用25毫升的滴定管,增大了读数误差,使结果超出合格限(当然同时还有其它的误差因素存在)。相反,校准EDTA标准溶液的浓度时,由于系用10毫升滴定管,需要滴约15毫升EDTA溶液,这样前后共有四次读数,导致读数误差增大。又如滴定异烟肼时,由于硫代硫酸钠的浓度低,(0.02moL)用50毫升滴定管共滴定约110毫升,由于读数误差(加上其它的因素),使测得含量95%。改用O.05moL的硫代硫酸钠溶液滴定。该检品又可合格。总之,化验师应当重视这些最基本的问题,以减少误差。二、准标溶液的浓度应定期校准标准溶液,某些标准溶液是比较稳疋的。下表是几种常用标准溶液短期内的浓度改变;标准溶液的浓度改变表(未改正体积)http://ng1.17img.cn/bbsfiles/images/2012/10/201210081113_395035_1611037_3.jpg上表可看出:常用的标准溶液在短期内浓度无明显改变。但应指出,标准溶液应当定期校准,作者认为每月应重新标定。标准溶液的体积可随室温改变而有改变。常用的一些标准溶液由200C升至300C,其体积可改变约为O.2一O.3%。应当注意:温度升高水要蒸发,冷凝于瓶子的内面空壁上,这对标准溶液的体积与浓度是有影响的,必须在用前摇匀。制备标准溶液,常须使用无二氧化碳的蒸馏水。蒸馏水新煮沸放冷,无二氧化碳,但注意应在无二氧化碳的空气下冷却(连接苏打石灰管)。碘的标准溶液据认为是不稳定的,但在下述条件下制备的碘标准溶液,每隔数周重标化一次,在一年内其浓度无任何改变。不管碘溶液的浓度,每升含KI应不少于40克;贮于有色玻塞瓶中并放在冷处,不能接触塑料、橡胶或软木塞;在使用中不受灰尘等沾污。取表面无油脂的纯碘经P2O5干燥24小时,准确称量,也可制备碘的标准溶液。硫代硫酸钠溶液的稳定性,多年来是可疑的。有研究指出,由于所用蒸馏水的酸度,可影响稳定。蒸馏水放在一般实验室环境下吸收空气中二氧化碳,其p H约为5~5.5,因此,在预备之前加氢氧化钠于蒸馏水中,调pH为7.O一7.5容器瓶应充分洗净,再以稀酚水洗涤,可杀灭细菌,并保证溶液稳定。重铬酸钾的标准溶液是稳定的。将K2Cr2O7在2000C干燥数小时(可磨成细粉后干燥),由准确称重计算浓度。也有推荐在110一1200C烘烤的。络合滴定用的EDTA常含杂质NTA(三价氮基三醋酸),分析纯EDTA中可达250ppm,它亦能与被测金属生成络合物,但络合能力仅弱于EDTA,此结果可影响终点。指示剂(如铬黑一T)溶液可能由于微量金属的催化作用,长期存放则氧化变质。在滴定时一些微量金属杂质自身可与指示剂或EDTA作用,则终点不明显,使滴定有很高的空白值(可加掩蔽剂如氟化物或氰化物消除干扰)。
特征形态液态 基体异辛烷主要分析方法气相色谱法,重量容量法规格1 mL/瓶用 途作为量值传递的标准,用于气相色谱-质谱仪(GC-MS)的校准和检定、分析方法评价,以及质量控制等方面。保存条件保存于干燥、洁净、避光的环境中使用注意事项最小取样量为1mL。六氯苯属于有害物质,使用时应注意防护,避免吸入或与皮肤接触,使用后剩余的溶液应进行专门的集中处理。
标准溶液稀释用的容量瓶,大家怎么清洁干燥?一般是25ml的
成分定量分析中,有些需要溶液煮沸才能溶解,对于熔点很高的溶液,大家怎么处理的,升温设施用的是什么
有学生问我一个问题:说上级专家指导说标准溶液的浓度只能有小数点后第四位,比如:0.00245mol/L的浓度就是不对的,只能是0.0024。理由是我们的衡器/量器达不到这个精度。老师觉得应该怎么处理呢?我当时就对他说:你能反抗权威不?坚持真理是需要底气的。你是自己没理解好有效数字。日常工作中接触到的标准溶液一般是容量分析用的滴定用标准溶液(好象日本是可以直接购买的),国家标准物质中心购买的溶液,还有就是化学对照品。1、滴定用标准溶液的有效数字滴定用标准溶液一般是标定浓度的,一般的公式:C=m/V/M,m为基准物质的质量,V为标定的体积(未考虑空白);M为基准物质的摩尔质量。按有效数字运算规则,滴定用标准溶液的有效数字一般4位。按GB/T 601规定,0.02mol/L以下浓度的溶液需用前稀释。稀释的有效数字也是乘除的运算规则,所以0.02mol/L以下浓度的溶液也应该有4位有效数字2、国家标准物质中心购买的溶液或化学对照品,比如金属标准溶液1mg/mL,证书上一般会有1%的不确定度,那这个溶液的浓度应该是1.00ml/mL,经过稀释,一般应该有3位有效数字。所以标准系列的有效数字应该这样写:0.00, 5.00, 10.0. 50.0, 100mg/L。对于如sigma对照品,一般是以99%来表示纯度的,按均匀分布其纯度是0.995+-0.0029,它也有三位有效数字。该对照品经称重,稀释得到的标准应用液浓度也是3位有效数字。
[align=center][b]标准溶液的分类及管理介绍[/b][/align]1[b]标准溶液的分类[/b]当我们以化学分析法或仪器分析法进行分析测定时,往往需要配置标准溶液。标准溶液就是已知其主体成分或其他特性量值的溶液。按照用途的不同,又分为滴定分析用标准溶液、杂质测定用标准溶液和pH测量用标准溶液。[b]1、标准滴定溶液[/b]滴定分析用标准溶液主要用于测定试样中主体成分或常量成分,有两种配制方法:一是用一级或二级标准物质(又称“基准试剂”)直接配制;二是用分析纯以上规格的试剂配成接近所需浓度的溶液,再用标准物质进行测定(称为“标定”)。GB601-88《滴定分析用标准溶液的制备》是我国唯一的标准方法。标准滴定溶液用物质的量浓度表示,符号为c(B),单位为molL-1,意指每升溶液中含有的滴定剂B为基本单元的物质的量(mol)。例如,某硫酸标准滴定溶液浓度为c(1/2H2SO4)=0.1000molL-1,又如,c(1/5KMnO4)=0.1021molL-1。[b]2、杂质测定用标准溶液[/b]杂质测定用标准溶液又称仪器分析用标准溶液,GB602-88《杂质测定用标准溶液的制备》给出了83种杂质标准溶液的制备方法。该种溶液所含的元素、离子、化合物或基团的量,以每毫升含有多少毫克表示。该标准规定的多数标准溶液浓度为0.1mgmL-1,少数是1mgmL-1,仅一种是10mgmL-1。规定浓度下溶液比较稳定,可称为贮备液,当需要使用更低浓度时可按要求稀释,制成标准系列溶液。[b]3、pH测量用标准溶液[/b]当用pH计测量溶液的酸度时,必须先用pH标准溶液对pH计进行校准。[align=left]2[b]标准溶液的管理[/b][/align]1、标准溶液的制备和标定、使用管理,由检验室设专人负责。2、标准溶液的制备必须严格按GB/T601-2002滴定分析(容量分析)用标准溶液的制备规定进行,专人配制和标定,且不得少于两人,标定时应详细记录标定过程。3、标准溶液实行标志管理,制备好的标准溶液应在标签上注明名称、浓度、基准物质名称、配制人、配制日期、标定人、标定日期、保存时间,并合理放置,由专人妥善保管。4、标准溶液的配制人和标定人要填写“标准溶液的制备与标定原始记录”。配制好的标准溶液由室主任批准后,方可使用。5、标准溶液在检验室内应单独放置,并保证室内环境条件符合要求;标定好的标准溶液在常温下的保存时间不得超过两个月;超过期限的标准溶液由配制人员重新标定,作好相应的记录和标签。6、检验人员使用标准溶液发现异常时,应及时向室主任反映,做好相应处理。转载于《分析测试百科网》
大家好,问一下比较菜的问题,我做有机有段时间了,对配置标准溶液做曲线的容器仍然困扰,首先如果用容量瓶配制,会消耗较多的有机溶剂,而且如果不是塑料塞子或者塞不紧,会漏液,因为有些有机溶剂粘度小于水,导致标准溶液配置不准。其次,如果用其他的没有刻度的安培瓶或者小试剂瓶,称量一定量的标准物质,或者量取一定量的标准溶液,用一定量的溶剂来稀释,这样的话可以节省了溶剂,但是定容体积会发生不准的情况。】因此希望咨询和请教一下大家,有没有什么好的意见学习一下,多谢了!
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
1标准溶液的配制方法及基准物质标准溶液是指已知准确浓度的溶液,它是滴定分析中进行定量计算的依据之一。不论采用何种滴定方法,都离不开标准溶液。因此,正确地配制标准溶液,确定其准确浓度,妥善地贮存标准溶液,都关系到滴定分析结果的准确性。配制标准溶液的方法一般有以下两种:1.1直接配制法用分析天平 准确地称取一定量的物质,溶于适量水后定量转入容量瓶 中,稀释至标线,定容并摇匀。根据溶质的质量和容量瓶 的体积计算该溶液的准确浓度。能用于直接配制标准溶液的物质,称为基准物质或基准试剂,它也是用来确定某一溶液准确浓度的标准物质。作为基准物质必须符合下列要求:(1) 试剂必须具有足够高的纯度,一般要求其纯度在99.9%以上,所含的杂质应不影响滴定反应的准确度。(2)物质的实际组成与它的化学式完全相符,若含有结晶水(如硼砂Na2B4O7.10H2O),其结晶水的数目也应与化学式完全相符。(3) 试剂应该稳定。例如,不易吸收空气中的水分和二氧化碳,不易被空气氧化,加热干燥时不易分解等。(4)试剂最好有较大的摩尔质量,这样可以减少称量误差。常用的基准物质有纯金属和某些纯化合物,如Cu, Zn, Al, Fe和K2Cr2O7 ,Na2CO3 , MgO , KBrO3 等,它们的含量一般在99.9%以上,甚至可达99.99% 。应注意,有些高纯试剂和光谱纯试剂虽然纯度很高,但只能说明其中杂质含量很低。由于可能含有组成不定的水分和气体杂质,使其组成与化学式不一定准确相符,致使主要成分的含量可能达不到99.9%,这时就不能用作基准物质。一些常用的基准物质及其应用范围列于表2.1中。表2.1 常用基准物质的干燥条件和应用基准物质干燥后的组成干燥条件℃标定对象名称化学式碳酸氢钠NaHCO3Na2CO3270─300酸十水合碳酸钠Na2CO3·10H2ONa2CO3·10H2O270─300酸硼砂Na2B4O7·10H2ONa2B4O7·10H2O放在装有NaCl和蔗糖饱和溶液的密闭器皿中酸二水合草酸H2C2O4·2H2OH2C2O4·2H2O室温空气干燥碱或KMnO4邻苯二甲酸氢钾KHC8H4O4KHC8H4O4110─120碱重铬酸钾K2Cr2O7K2Cr2O7140─150还原剂溴酸钾KBrO3KBrO3130还原剂草酸钠Na2C2O4Na2C2O4130KMnO4碳酸钙CaCO3CaCO3110EDTA锌ZnZn室温干燥器中保存EDTA氯化钠NaClNaCl500─600AgNO3硝酸银AgNO3AgNO3220─250氯化物1.2 间接配制法(标定法)需要用来配制标准溶液的许多试剂不能完全符合上述基准物质必备的条件,例如:NaOH极易吸收空气中的二氧化碳和水分,纯度不高;市售盐酸中HCl的准确含量难以确定,且易挥发;KMnO4和Na2S2O3等均不易提纯,且见光分解,在空气中不稳定等。因此这类试剂不能用直接法配制标准溶液,只能用间接法配制,即先配制成接近于所需浓度的溶液,然后用基准物质(或另一种物质的标准溶液)来测定其准确浓度。这种确定其准确浓度的操作称为标定。例如欲配制0.1mol.L-1HCl标准溶液,先用一定量的浓HCl加水稀释,配制成浓度约为0.1mol?L-1的稀溶液,然后用该溶液滴定经准确称量的无水Na2CO3 基准物质,直至两者定量反应完全,再根据滴定中消耗HCl溶液的体积和无水Na2CO3 的质量,计算出HCl溶液的准确浓度。大多数标准溶液的准确浓度是通过标定的方法确定的。在常量组分的测定中,标准溶液的浓度大致范围为0.01 mol.L-1至1 mol.L-1,通常根据待测组分含量的高低来选择标准溶液浓度的大小。为了提高标定的准确度,标定时应注意以下几点:⑴ 标定应平行测定3- 4次,至少重复三次,并要求测定结果的相对偏差不大于0.2% 。⑵ 为了减少测量误差 ,称取基准物质的量不应太少,最少应称取0.2g以上;同样滴定到终点时消耗标准溶液的体积也不能太小,最好在于20mL以上。⑶ 配制和标定溶液时使用的量器,如滴定管 ,容量瓶和移液管 等,在必要时应校正其体积,并考虑温度的影响。⑷ 标定好的标准溶液应该妥善保存,避免因水分蒸发而使溶液浓度发生变化;有些不够稳定,如见光易分解的AgNO3和 KMnO4等标准溶液应贮存于棕色瓶中,并置于暗处保存;能吸收空气中二氧化碳并对玻璃有腐蚀作用的强碱溶液,最好装在塑料瓶 中,并在瓶口处装一碱石灰管,以吸收空气中的二氧化碳和水。对不稳定的标准溶液,久置后,在使用前还需重新标定其浓度。2 标准溶液浓度表示方法2.1 物质的量浓度(c,简称浓度)物质的量浓度是指单位体积溶液中含溶质B的物质的量,以符号cB表示。即cB =nB /VB (2.1)因 nB = mB/MB (2.2)所以 mB=cBVBMB (2.3)式 (2-1)中,B代表溶质的化学式,nB为溶质B的物质的量,它的SI单位是mol,VB表示溶液的体积,其SI单位是m3;所以物质的量浓度cB的SI单位是mol.m-3,在分析化学中常用的单位为mol.L-1或mol.dm-3 。在式(2-2)中,mB是物质B的质量,常用单位
溶液怎样稀释分析化学,特别是精密的仪器分析,其检测范围往往是ppm\甚至是ppb级,这个就需要我们配置ppm甚至是ppb级的标准溶液。怎样配置标准溶液?这个问题是个问题。我要配置1ppb的铅标准溶液,国家标准物质为1g/L,首先吸取1毫升标准液于100毫升容量瓶中,加水定容,此时该储备液浓度为10mg/L,随后吸取该储备液1毫升,再次稀释到100毫升容量瓶中,此时的储备液浓度为0.1mg/L,随后吸取二次储备液0.25毫升,加入25毫升比色管中。此时的标准溶液浓度就是1ppb。此方法为逐级稀释的办法,该方法稳定性好,缺点是操作繁琐。第二种办法就是用移液枪小体积稀释,量取0.1微升的标准溶液,随后将其转移到100毫升的容量瓶中,加水定容。该方法简单快速,缺点是系统误差大,往往导致结果不准确。仪器分析每一次的移液误差传递到下一步将会从绝对误差转变为相对误差。在实际工作中,可以利用微量注射器或者移液枪,将1微升的液体稀释到1毫升的样品瓶中,或者1毫升的小离心管中,这样就可以稀释1000倍了。配置标准溶液的时候,不同的人有不同的爱好。有的人喜欢用移液枪,还有的人喜欢用移液管,方法不同,但是原理相同。一般来说移液管经过计量校准以后,其溯源性要好于移液枪。但是移液枪比移液管方便许多。我推荐还是使用移液管。有的人喜欢用容量瓶配置标准溶液,而有的人喜欢用比色管配置标准溶液。早些天去省局学习,一位做离子色谱的大哥,竟然对我说,他不知道比色管是什么东西,顿时我傻眼了!配置溶液的时候,有的人喜欢在小烧杯里溶解,随后转移到容量瓶中,然后反复清洗转移。这样做符合分析化学的基本原理,其缺点就是增加的一步过程中,容易引进污染;有的人喜欢直接将标准品放入容量瓶中,然后直接稀释,这样做方便,也不容易引进污染,其缺点就是容量瓶瓶口太小,容易撒到外面去。每一次转移和定容的过程中,都要把容量瓶摇匀,这样才能保证您的标准曲线做到4个9哦!今天您做到了吗?
购买了计量科学研究院的水中甲醛溶液标准物质(BW3450),证书上特性量值栏给出的标准值是11.0mg/mL,包装栏写的是本标准物质采用玻璃安瓿瓶包装,2mL/支。请教大家一下,我想配制一定浓度的甲醛标准溶液,有2种方案,方案一是:将标液物质在室温下平衡,然后打开安瓿瓶,用已经检定的移液管或是[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]移取,在确保准确吸取的条件下尽量多吸取一定体积的V(mL),记下该体积,转移到容量瓶中,比如是检定合格的25mL棕色容量瓶,加水稀释定容,计算甲醛的溶液浓度=(11.0mg/mL*VmL)/25mL即得。方案二:将标液物质在室温下平衡,然后打开安瓿瓶,将安瓿瓶中的甲醛标准溶液全部转移到检定合格的25mL棕色容量瓶,用少量水洗涤安瓿瓶5次以上,将每次洗涤的溶液也转移到容量瓶中,加水稀释,定容至刻度,计算甲醛溶液浓度=(11.0mg/mL*2mL)/25mL即可。请问大家觉得那种方法正确??平时大家是怎么配制的?另外我发现证书上特性量值栏(和标明的标准值11.0mg/mL同一栏)相对扩展不确定度(%)(K=2)为3,这个不确定度应该是浓度的不确定度吧,没有包括2mL体积在内吧? 既然不包括2mL这个体积在内,那么配制标液的时候我觉得还是用第一种方案好。以第一种方案可以用移液管引入的不确定度、甲醛标准溶液物质的不确定度和容量瓶等引入的不确定度合成最后配制成甲醛标液的不确定度~~~~
刚开始做化学分析,以前是玩生物的。例如:滴定氯化钠用的0.1mol硝酸银标准液一定要用2级标准品配制吗?配制完了还得用基准氯化钠标定。那么硝酸银标准液能用一般的AR硝酸银配制吗?然后用基准氯化钠标定
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
假设我配制错了,直接把容量瓶里的标准溶液倒了然后用自来水冲洗3-5边,纯净水冲洗3边,应该可以继续使用了吧?
请问水中氟成分分析标准物质(例如[url=https://www.bzwz.com/p_55/p_1218.html]GBW(E)083222[/url]) 水中氟溶液标准物质(例如[url=https://www.bzwz.com/p_55/p_128254.html]GBW(E)083311[/url]),都是北方伟业生产的,但该两类产品有何区别?
全面详解常用标准溶液的配制与标定(分析与实验)标准溶液是援引美国加联数据库定义已知准确浓度的溶液。在滴定分析中常用作滴定剂。在其他的分析方法中用标准溶液绘制工作曲线或作计算标准。如果试剂符合基准物质的要求 (组成与化学式相符、纯度高、稳定),可以直接配制标准溶液,即准确称出适量的基准物质,溶解后配制在一定体积的容量瓶内。如果试剂不符合基准物质的要求,则先配成近似于所需浓度的溶液,然后再用基准物质准确地测定其浓度,这个过程称为溶液的标定。标准溶液的配制方法直接配制法:用基准物质(如K2Cr2O7)来直接配制标准溶液。因为这些基准物质能够符合基准物质条件,容易得到。此法溶液浓度由计算而得,浓度不必标定。间接配制法:不能得到符合基准物条件的物质(如NaOH 、HCl ),先配制近似浓度的标准溶液,再用基准物质或另一种已知浓度的标准溶液来标定它的浓度。这种配制方法比较费事,但大多数标准溶液都是这样配制的。溴酸钾标准溶液的配制与标定配制:c(1/6KBrO3)=0.1mol/l称取3g溴酸钾,溶于1000ml水中,摇匀标定方法及计算同溴标准溶液的标定及计算碘标准溶液的配制与标定配制:c(1/2I2)=0.1mol/l称取13g碘及25g碘化钾,溶于100ml水中,稀释至1000ml,摇匀,保存于棕色具塞瓶中。标定:称取0.15g预先在硫酸干燥器中干燥至恒重的基准三氧化二砷,称准至0.0001g。置于碘量瓶中,加4ml氢氧化钠溶液溶解,加50ml水,加2滴酚酞指示剂,用硫酸溶液中和,加3g碳酸氢钠及3ml5g/l淀粉指示剂,用配好的碘溶液c(1/2I2)=0.1mol/l滴定至溶液呈浅蓝色。同时作空白试验。
各位大侠,因为工作中要配制很多种标准溶液,我们用配制过标准溶液的容量瓶配制其它标准溶液时,进样后一直有很大干扰,不知各位有什么好的清洗方法没有
[size=4][B]关于标准溶液配制的问题-重量法和容量法?的总结。[/B][/size]原帖链接: http://www.instrument.com.cn/bbs/shtml/20080705/1341908/前段时间版面的帖子“关于标准溶液配制的问题-重量法和容量法?”经过大家的讨论,现已结贴。在目前的农残检测分析中,定量方法一般都是相对定量的方法,因此标准溶液的配制是一个重要的环节,一般常用的标准溶液配制方法都是使用的体积法,既然是使用普遍,一定有其自身的适用性。大家看以下的阐述,或许能清晰一些:[B]calfstone:[/B]个人观点,与同仁探讨。体积法和重量法大家都会在实验中碰到。到底哪个好,哪个更适用于你的分析是首先考虑的问题。其实问题指向的肯定是哪个带来的误差小,不确定度小(我一直定义不确定度为可操作/可量化的误差)1、先探讨哪个好的问题。举一个简单的例子来说明。配制1mg/ml 或 1mg/g NaCl 溶液(前者是体积法后者是重量法),由于分子都一样,主要的不确定度来至分母(1ml和1g).对于体积法,按JJG196-1990规定:A级的1ml单标线容量瓶的容量允差为0.010ml,相对不确定度为0.01/qrt(3)=0.058 对于重量法,普通的万分之一天平,简单的允差0.1mg,按正态分布的B类评定,其相对不确定度为0.000033。两种方法配制的溶液按不确定度表示方法分别为:体积法:1+-0.058mg/ml 重量法:1+-0.000033mg/g(忽略分子的不确定度)。很明显重量法的不确定度要小很多。2、再来探讨下适用性问题。体积法对温度的要求非常严格,JJG196-1990检定规程中限定室内温度在20+-5摄氏度,且水温和室稳之差不超过2摄氏度。常规实验室操作很难限定如此严格的范围。虽然表述了很多体积法的不足,但我们做分析不是只停留在配制溶液这一个环节,还有整个分析过程。而整个分析过程中其它环节所引入的不确定度要比溶液配制要大很多(即使是体积法),这些环节主要有:样品重复性,标准曲线,仪器重复性等等。因此,站在分析全局的高度讲,去优化一个微小的不确定度来源(体积法变为重量法)对整个分析的不确定度是没有任何改善的。所以,采用体积法是很明智的选择。但如果你采用不好的配制溶液的策略,比如我的1中的举例的话,5%的相对不确定度还是很可观的了(我想没人用1ml的容量瓶来配制1mg/ml的溶液)。P.S. 听我一个到荷兰进修的同事讲,他们配制溶液都采用重量法。

原子吸收光谱分析中标准溶液配制 标准溶液是原子吸收光谱法测定样品中待测元素含量时必须用到的,下面就标准溶液的配制做以下阐述。1、 自行配制标准溶液(母液)必须采用基准物质,通常各元素合适的盐类来配置标准溶液,当没有合适的盐类可供使用时,可用相应的高纯金属丝、棒、屑。通常不使用海绵状金属或金属粉末,因为这两种状态的金属易引入污染物或容易氧化,纯度达不到要求。金属在使用前,一定要注意用酸清洗或者用砂纸打光,除去表面的污染物和氧化层。http://ng1.17img.cn/bbsfiles/images/2012/12/201212261343_415610_2352694_3.jpg举例:锌标准溶液的配制方法(1)、称取1.0000g金属锌(除去表面氧化膜)于300ml烧杯中,加入30-40ml盐酸(1:1),使其完全溶解后,加热煮沸几分钟,冷却后移入1000ml容量瓶中,以水稀释至刻度,摇匀。此溶液1ml含有1mg锌;(2)、称取1.2447g氧化锌(预先在900度灼烧至恒重),于300ml烧杯中,加入20ml硫酸(0.05mol/l),使其完全溶解后移入1000ml容量瓶中,以水稀释至刻度,摇匀。此溶液1ml含有1mg锌。自行配制,感觉误差比较大,一般配制好之后,要进行标定。2、 采用国家认可单位(如国家标准物质中心等)生产的商品1000ug/ml或500ug/ml的有证标准溶液。分取逐步稀释(一般稀释10-20倍,否则误差比较大)。http://ng1.17img.cn/bbsfiles/images/2012/12/201212261343_415611_2352694_3.jpg购买成品,比较方便,可以直接使用,有证书,质量可以保证。3、标准贮备溶液、标准工作系列溶液必须用超纯水或蒸馏水配置。标准工作系列保存不要超过一周(给一定的酸度,保存时间可以长一些),浓度很低的标准溶液(1ug/ml以下)使用时间最好不要超过2天;母液的保存时间视不同元素稳定性而定,通常为6个月-1年。标准溶液浓度的变化速度与标准溶液本身元素的性质、浓度、介质、容器、保存条件均有关系。(经常使用某一标液,如果实验没有特殊要求,建议给较大酸度,可以长期使用,使用时间的长短,可以观察使用过程中标液吸光度的变化,确定是否失效) http://ng1.17img.cn/bbsfiles/images/2012/12/201212261344_415612_2352694_3.jpg 4、 保存标准溶液的容器材质要根据不同元素及介质而定。无机贮备溶液置于聚四氟乙烯容器中,保持必要酸度(10%左右),保存在清洁、低温、阴暗的地方。容器必须洗净,对于不同容器采取的洗涤方法不同,通常将容器浸泡在5%的硝酸或盐酸溶液中。标准溶液要有专人保管,不允许随便乱拿乱放。5、 标准溶液(贮备溶液、标准工作系列)要标明溶液编号、名称、浓度、介质、配置日期、配置人。http://ng1.17img.cn/bbsfiles/images/2012/12/201212261345_415613_2352694_3.jpg标准溶液广泛使用于分析工作中,标准溶液浓度是否准确,必将影响实验结果的准确度,在配置、保存、使用标准溶液时,我们一定要慎之又慎!
在做食品接触材料试验时,按照GB/T 5009.69—2003食品罐头内壁环氧酚醛涂料卫生标准的分析方法测定酚含量时配制酚标准溶液方法如下:准确称取新蒸182℃~184℃馏程的苯酚约1 g,溶于水中移入1 000 mL容量瓶,加水稀释至刻度。请问1.新蒸182℃~184℃馏程的苯酚1g,这个馏程是指在苯酚在182℃~184℃蒸馏下得到的苯酚吗?在蒸馏过程中应注意一些什么问题?用什么样的蒸馏装置较合适?2.市场上有这种苯酚的标准溶液卖吗?就是苯酚标准溶液基体是水,不是其它物质3.可以把苯酚的标准溶液的基体换成别的物质吗?对实验结果会不会与有影响?
标准溶液是指已经准确知道质量浓度(或物质的量浓度)的用来进行滴定的溶液。一般有两种配制方法,即直接法和间接法3.2.1 直接法 根据所需要的质量浓度(或物质的量浓度),准确称取一定量的物质,经溶解后,定量转移至容量瓶中并稀释至刻度,通过计算即得出标准溶液准确的质量浓度(或物质的量浓度)。这种溶液也称基准溶液(Standard Solution)。用来配制这种溶液的物质称为基准物质(StandardSubstance)。对基准物质的要求是: ① 纯度高,杂质的质量分数低于 0.02%,易制备和提纯; ② 组成(包括结晶水)与化学式准确相符; ③ 性质稳定,不分解,不吸嘲,不吸收大气中C02,不失结晶水等; ④ 有较大的摩尔质量,以减小称量的相对误差3.2.2 间接法(也称标定法)若欲配制标准溶液的试剂不是基准物,就不能用直接法配制。(注意:优级纯或分析纯试剂的纯度虽高,但组成不一定就与化学式相符,不一定能作为基准物使用。)间接配制法是先粗配成近似所需质量浓度(或浓度)的溶液,然后用基准物通过滴定的方法确定己配溶液的准确质量浓度(或浓度),这一过程称为标定。
[color=#6495ED][color=#DC143C]请大虾帮忙推荐几家做化学滴定分析用的标准溶液!国内的就好,关键是要有国家标准物质定级证书,还有要便宜!当然买回来后,我们也会定期做做标定的!目前我就知道博林达算是比较大的。我们公司常用到0.1M Na2S2O3,0.1M I2,KIO3等等。先谢谢大家了[/color][/color]
用容量瓶盛装标准溶液、储备液、内标液是不是合理。
[align=center]关于容量分析过程中误差来源[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]化工室:姚文艳[/align] 在实验分析过程中,我们会严格遵循标准,同时也会有创新,但分析结果与真实值之间总会存在误差,而实验过程中的误差可以分为两大类:系统误差、偶然误差和过失误差。 一.系统误差:由于测定过程中某些固定的原因造成的,它可以通过对照实验,空白实验,仪器校准来消除。同时它也被称为可测误差。产生的原因有以下几点:1.方法误差 测定的方法本身所造成的。检验方法是否合理,所涉及的一切是否配套。滴定的分析过程中,溶液PH的调节,滴定终点与化学计量点之间不符合,反应过程中各类副反应都会是测定的结果存在高低差。如:重量分析中被测组分沉淀完全是不可能的,必须通过其他方法对溶解损失进行校正。2.仪器误差 仪器本身未经校准或仪器本身不合格。这就是为什么新领来的仪器需要校准,如天平、砝码、滴定管、以及各类容量器皿。就拿新买来的滴定管而言,我们先要对看滴定管刻线是否平整、明显,进而对它校准,采用的方法就是水在20℃密度与质量之间的关系求出校正值,之后方能开始使用。而一些容量器皿最重要的是对其气密性把控。3.试剂误差 一方面由于蒸馏水含有杂质,另一方面试剂的过期变质,沉淀都会造成误差。不同的试剂存放条件:有的试剂需要避光保存例如高锰酸钾,碘标液;存放温度,对于溶液的温度存放不同,环境温度的不同,我们可以通过溶液不同温度之间的相关性对它进行校正,在计算过程中通过加入温度校正值减小因为存条件而引起的误差;配制过程中溶剂是否都是需要注意的,有的指示剂选择乙醇作为溶剂,而有的会选择丙酮做溶剂,不能一味的盲目的只认水。4.操作误差 这个误差主要来源于分析工作者,对于条件不熟练,颜色敏锐程度以及固有的一些习惯,他们自己的主观原因。对于实验条件我们可以一步步参考标准,但过程的把控每个人和每个人都是不一的,有的人对颜色变化敏锐,有的人迟钝;滴定管读数偏高或偏低都会造成误差产生。 二.偶然误差又称随机误差,它是由于外界的各种不受控制的物理原因造成的,测量过程中的温度、湿度、气压波动、仪器性能变化等,不可控的,随机的,它不可测量也无法消除。虽然它无规律但也服存正态分布。 三,过失误差 分析过程中由于自己的粗心过失造成的误差,这种事情在工作中很常见,但它都不属于应该有的,这就是一个人,一名化验员的质量意识,我们必须加强自身的责任感,该认真的时候必须一丝不苟,严格遵循操作规程,误差较大的要舍弃,过失误差是完全可以避免的。数据的准确性离不开好多的东西,我们应该在能遵循规程切不断创新的的前提下严格要求自己,做到精益求精!







 我要推广仪器
我要推广仪器
 下载APP
下载APP