413家检测机构认可资格被注销
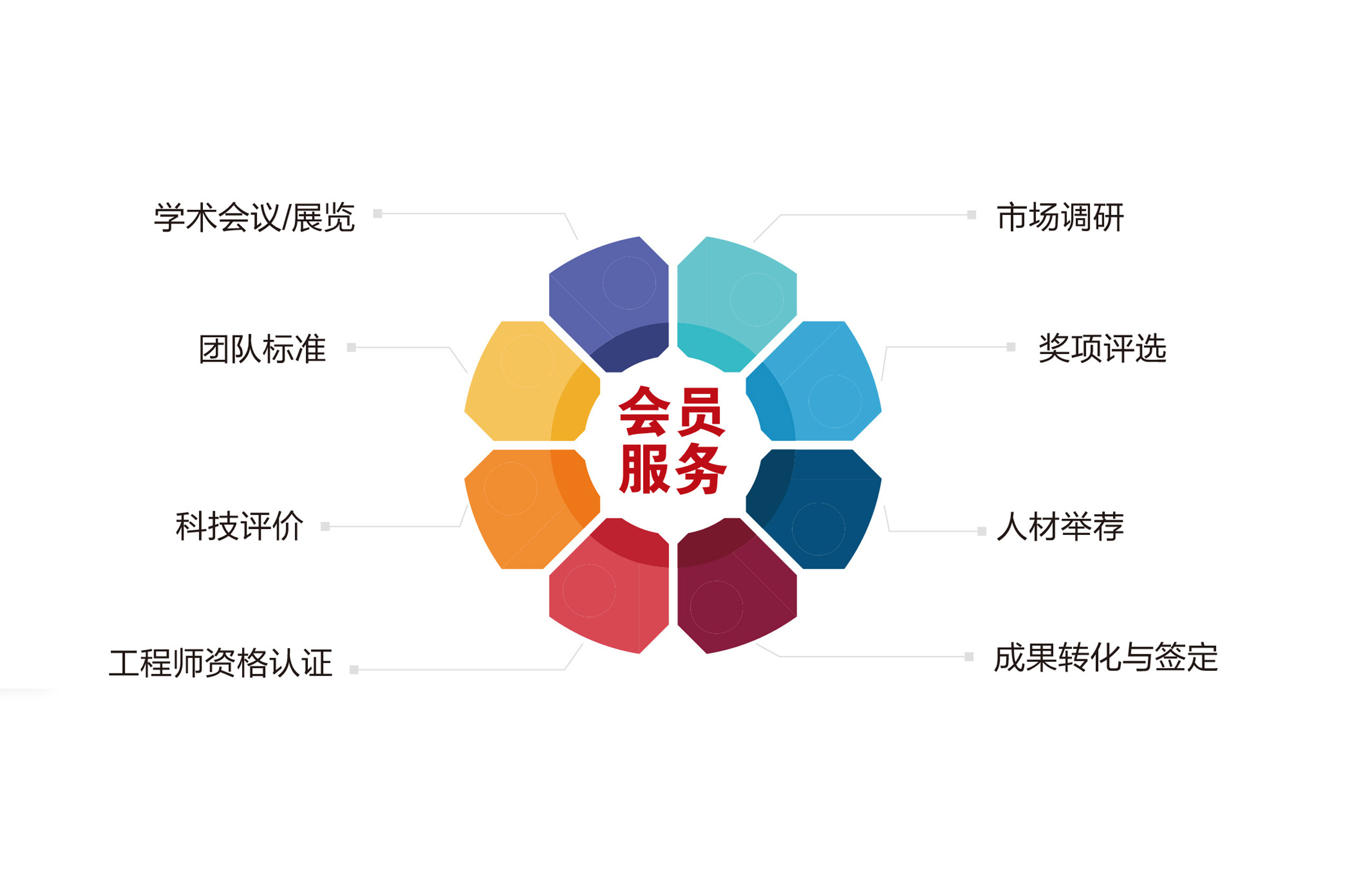
法制网-法制日报(北京)8月22日讯 记者今天从中国合格评定国家认可委员会获悉,该委坚持严格认可,截至2012年6月底,累计暂停了626家认证机构、实验室或检查机构的认可资格,累计撤销了327家认可资格,注销了413家认可资格。 2002年2月11日,国务院办公厅下发《关于加强认证认可工作的通知》,明确建立全国统一的国家认可制度。同年8月20日,我国统一的认可机构法律实体——中国合格评定国家认可中心成立。 据了解,10年来,我国认可制度领域拓宽了3倍,认可数量增长了5倍,认可的认证数量增长了8倍,认可业务取得了长足的进步。我国认可制度的齐全性及新兴认可制度的发展位居国际同行前列,覆盖了国际通行的认证机构、实验室、检查机构3大门类,包含了11项基本认可制度,具体有29个分项认可制度,包括:质量、环境、职业健康安全、食品安全、信息安全等管理体系认证和产品认证等。 截至今年6月底,我国认可的各类合格评定机构5500多家,认可范围内有效认证证书60多万张,连续9年稳居国际同行前列。我国认可的检测报告和认证证书已成为很多国家进口相关产品的有效凭证,可直接通关,免于再次检测。 10年来,我国已有30余部法律、法规和行政规章等行政规定直接或间接涉及与认可相关的规定。认可已成为强制性产品认证、有机产品认证、良好农业规范认证、食品安全管理体系认证、体育服务认证、生物安全三级和四级实验室等相关合格评定机构技术与管理能力的必备条件,成为质量、环境、职业健康安全、信息安全等管理体系认证技术与管理能力建立信任的基本方式 从事有关司法鉴定、工业产品质量控制和技术评价的实验室须获得计量认证或实验室认可。10年来,认可委员会还坚持国际化认可发展战略,积极推进国际互认,实现互认范围全覆盖。















 我要推广仪器
我要推广仪器
 下载APP
下载APP