分析方法验证:在制药行业中采用TOC方法进行清洁验证
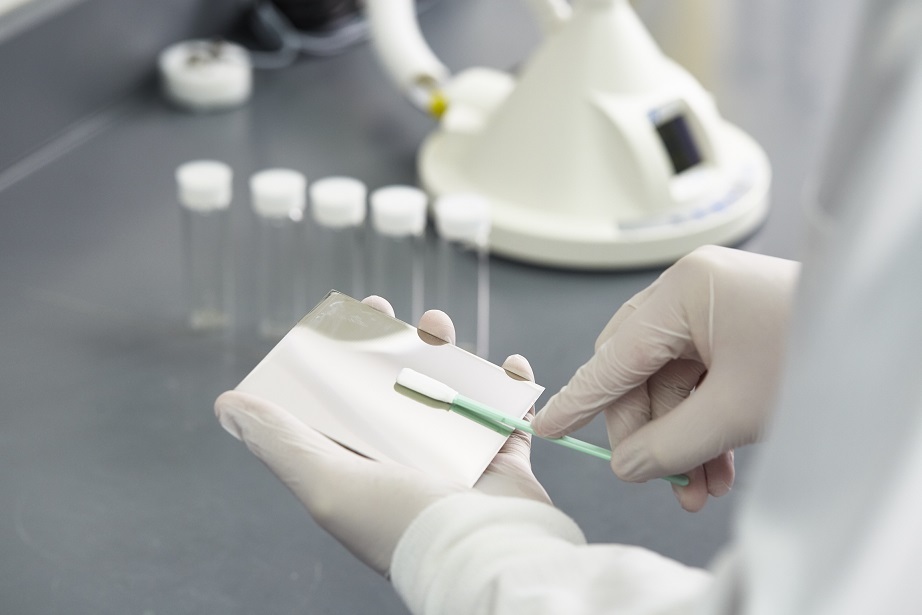
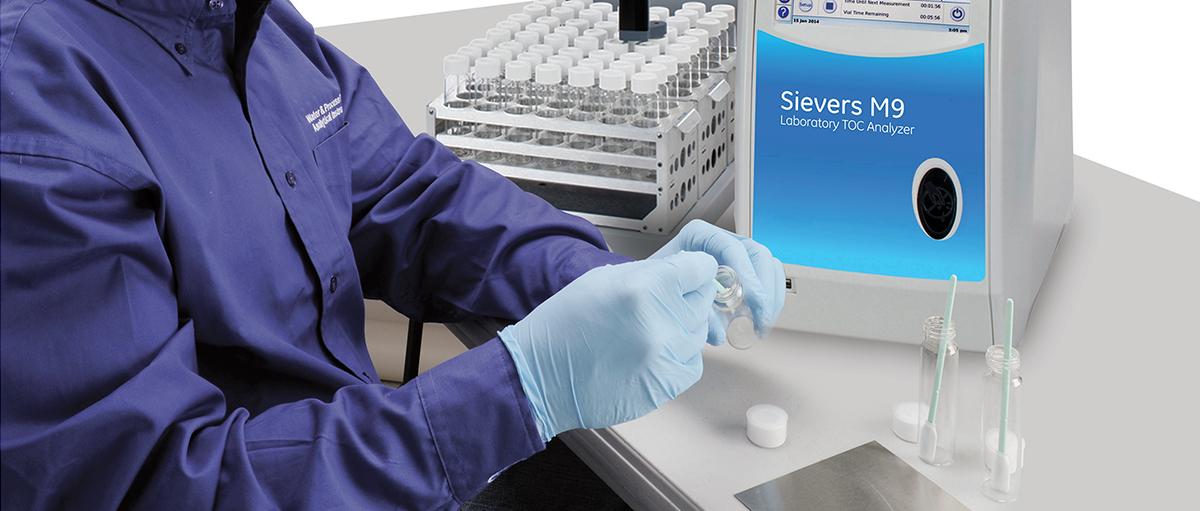
寻求改进质量和提高效率的药品生产商对使用Sievers® 总有机碳(TOC)分析仪进行清洁验证的兴趣越来越浓。大多数制药或生物科技厂家目前都配有TOC分析仪以符合美国药典USP、中国药典ChP的水检测要求,以放行纯化水或注射用水用于清洁或生产过程。因此,大多数厂家已经拥有用于清洁验证的TOC测定方法。TOC是FDA认可的一种方法①,用于评估所给样品中所有含碳的化合物,以确保所有设备的清洁都符合所建立的清洁标准。TOC分析允许开发一种方法,用于检测由化合物、分析物或残留物通过直接(擦拭)或间接(冲洗)取样而形成的碳浓度。潜在目标残留物包括药物活性成分(API)、药品赋形剂、蛋白质、蛋白质副产品和清洁剂或成分。1996年,国际协调会议(ICH)在FDA(CDER & CBER②)的协助下,创建了指导文件《Q2B:分析步骤的验证》。该文档的目的是为制药公司如何考虑清洁验证分析程序的各种验证特征提供参考。本文提供了与下列参数相关的多个实例,这些实例均与TOC方法验证有关,因而此应用说明呼应了Q2B指导文件:检出限和定量限确定分析物的准确度和精确度线性和回收百分比分析方法的稳固性③检出限和定量限检出限(LOD)用于评估何时信号是仪器噪音的结果还是化合物的反应。LOD被视为样本中分析物的最低检测量,但没有必要的足够的统计确定性来定量。定量限(LOQ)是对数据有意义还是无意义提供指导而建立的值。低于LOQ的仪器反应表示存在有机物,但无法定量实际浓度。分析仪中的读数高于已建立的LOQ则被视为可定量或有意义的数据。为了确定背景TOC的浓度并推导出用于清洁验证方案的LOD和LOQ,必须准备低TOC的水空白或棉签空白(如果适用)来计算实验中水和小瓶的碳成分。一旦已经从这些样本中确定了标准偏差,则通常是将标准偏差分别乘以3和10来获得LOD和LOQ④。确定分析物的准确度和精确度了解TOC分析方法验证中准确度和精确度的区别非常重要。准确度与测得值和分析物的真实值的接近程度相关。通常,准确度是计算仪器验证时测得的标准品的TOC浓度与预期的标准品TOC浓度的差值百分比(即+7%)所得。精确度通过标准偏差或RSD(相对标准偏差)度量。精确度与所给样本的多个分析结果相互之间的接近程度相关。在TOC方法验证期间,通过分析加了(添加)已知浓度的目标残留物的样品可以测定准确度和精确度,并可以评定差值百分比和RSD。ICH文件推荐至少在三个浓度级别上至少进行九次测定来评估准确度和精确度,这三个浓度级别涵盖了仪器的指定范围⑤。线性和回收百分比验证通常,线性测试校验仪器反应值是否与所研究分析物的浓度具有线性关系。图1演示了TOC浓度范围从1.00 ppm到7.50 ppm,牛血清白蛋白(BSA)的线性关系,其中含低TOC水的小瓶中加了已知浓度的BSA。这个例子演示了理论浓度(x轴)对所测得的浓度(y轴)作图所得到的两者之间的线性关系,y=(m)x+b。分析仪的反应值与所研究化合物的相关系数(R² )应大于0.97。图1. 数据使用Sievers实验室TOC分析仪获得为了确定TOC方法用于分析目标残留物的适用性,有必要确定分析方法可达到的回收率。以下例子使用CIP-100制备已知TOC度的溶液,并将已知量的样本放到不锈钢片上,演示了直接取样方法。在BSA的例子中,在不锈钢片上添加三个递增浓度的CIP-100清洁液,擦拭不锈钢片,然后将此棉签放到已知量的低TOC水中。表1提供了从不锈钢片表面获得的回收百分比结果。分析方法的稳固性与实际回收率同样重要的是,用于确定所研究化合物回收百分比的TOC分析方法的重现性或稳健性。在清洁验证方法开发中稳固性是指结果不受方法中参数、或样本之间的小而微妙的变化的影响的能力。还提供了正常使用期间的可靠性指示(例如各个分析员的取样方法)。若希望得到高回收率,回收率一直保持可重复性也同等重要或更为重要,并在整个方法开发期间一直需要对回收率进行检测。表1和表2提供了CIP-100棉签回收率分析信息,由两个不同的分析员测试样本间的变化。要考虑的最后几点评估制药产品质量水平的测试步骤要遵从各项要求。具体到清洁验证来说,当前的药品生产质量管理规范[21 CFR 211.194(a)] 要求,用于评估药品是否符合已建立规范的测试方法必须满足准确度和可靠性的合适标准⑦。同时考虑到分析方法的验证是通过实验室研究建立的过程,本应用说明中说明的(TOC)方法的性能特征满足计划进行的分析应用的某些要求,例如符合药典的水排放和清洁验证。参考文献FDA网站:www.fda.gov/cder/guidance/cGMPs/equipmenthtm。药品评估与研究中心(CDER)和生物制品评估和研究中心(CBER)。Guidance for industry Q2B: Validation of Analytical Procedures. Methodology. November 1996. ICH, FDA, CDER, CBER.Taylor, John K. Quality Assurance of Chemical Measurements. Lewis Publishers imprint of CRC Press 1987.USP Validation of Compendial Methods.The Swab Recovery Determination of CIP-100 in Solutions by TOC Analysis Using a Sievers TOC Analyzer, Steris Corporation Analytical Method 1993. 7. 21 CFR 211.194(a) Laboratory Records.21 CFR 211.194(a) Laboratory Records.◆ ◆ ◆联系我们,了解更多!

























 我要推广仪器
我要推广仪器
 下载APP
下载APP