2018年56款FDA新药深度盘点
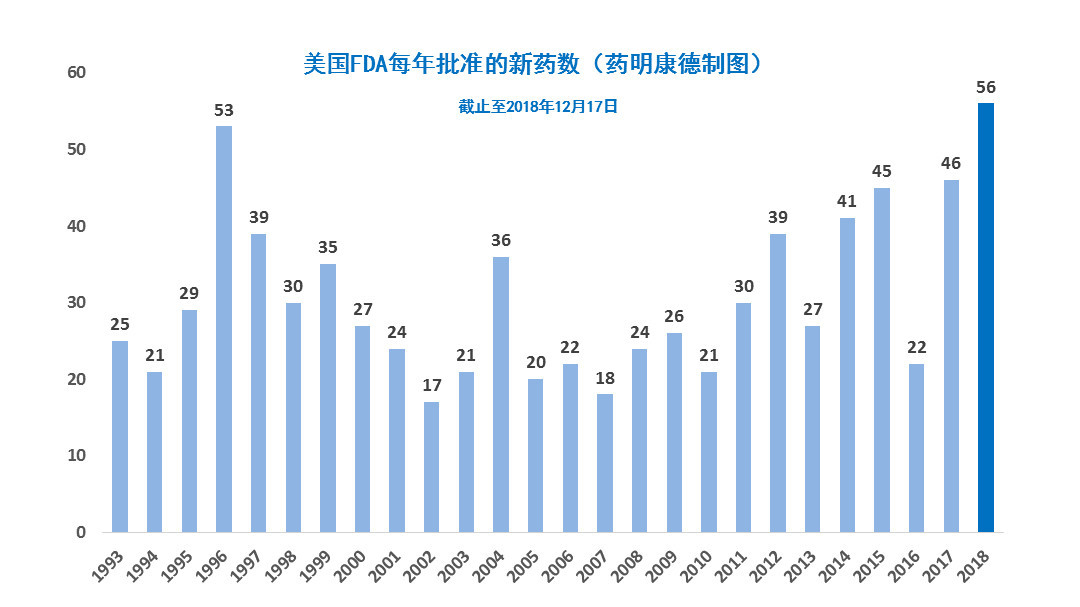
p 2018年是不平凡的一年。截止到发稿时,美国FDA在今年一共批准了56款新药。这个数字不但打破了尘封20年的纪录,更是创下了历史新高。本文对这56款新药做一个盘点。 br/ /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/445ad703-9c08-4f02-b003-86b2373a71a3.jpg" title=" 1.jpg" alt=" 1.jpg" / /p p span style=" color: rgb(112, 48, 160) " span style=" color: rgb(0, 112, 192) " strong 罕见病的胜利 /strong /span /span /p p 每年的获批新药中,抗癌疗法永远是大头。据公开信息统计,在过去的5年里,癌症疗法少则占获批新药比例的20%,多则超过30%,是最常获批的新药类型。今年,总共有15款创新抗癌疗法获批上市,占比27%。这两个数字分别是过去5年的最高和第二高,表现依旧亮眼。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/b0a8efdf-6b8f-4649-8fda-68f3ec4d0297.jpg" title=" 2.jpg" alt=" 2.jpg" / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " ▲美国FDA今年批准的新药中,有15款抗癌疗法 /span /p p 但在2018年,获批新药的数目却首次未由抗癌疗法所主导。今年,有31款治疗罕见疾病的新药获批,占比55%,创下了历史新高。美国CDER的年度报告也指出,今年的创新分子实体中,大部分是治疗罕见疾病的孤儿药,这是历史上的头一遭。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/c2a6bd70-ea1c-407a-ac93-2032e7a83438.jpg" title=" 3.png" alt=" 3.png" width=" 412" height=" 388" style=" width: 412px height: 388px " / /p p 如果我们往回看,就会发现这样的结果不是偶然。早在2017年初,就有不少分析专家指出,新上任的FDA局长Scott Gottlieb博士对罕见病有足够的重视,并希望FDA能精简药物的开发流程,而不是设置障碍。而数字也证实了这一点。在2012-2016年这5年里,美国FDA平均每年批准17款罕见病新药。而在2017-2018年期间,这一平均数上升到了25,增幅接近50%。 /p p span style=" color: rgb(0, 112, 192) " strong 新药如何加速获批? /strong /span /p p Gottlieb博士上任后的第一场FDA全体会议,确立了“以患者为中心,立足于科学”的方针,让更多患者能够用上更多安全有效的新药。从结果上看,这两年的获批新药数也的确出现井喷,两年的获批新药总数已经超过了100款,与2016年的22款新药形成鲜明对比。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/8534e90b-73c3-438d-a950-da660be948a9.jpg" title=" 4.jpg" alt=" 4.jpg" width=" 495" height=" 439" style=" width: 495px height: 439px " / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " ▲最近两年获批的新药中,不少通过了FDA的四大加速政策 /span /p p 在短时间内能有那么多的新药获批,与FDA的四大加速政策(即突破性疗法认定,优先审评资格,快速通道资格,加速批准)是分不开的。我们看到,最近两年里,除了“加速批准”外,以其余叁大加速政策获批上市的新药,均创下了一个又一个的纪录。其中,今年每四款新药里,就有叁款通过“优先审评”上市。而获得“快速通道”的新药数,也从去年的18款勐增到目前的24款,增长超过3成。 /p p 综合来看,今年通过某一种加速政策上市的新药,共有42款,占总体的75%。 /p p span style=" color: rgb(0, 112, 192) " strong 多款创新疗法问世 /strong /span /p p 在今年批准的56款新药中,不乏创新疗法的影子。今年8月,Alnylam Pharmaceuticals带来了史上第一款RNAi疗法Onpattro。这也是RNAi技术在2006年斩获诺贝尔生理学或医学奖之后,等待12年所取得的另一项荣光。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/f63f0e8a-8181-4859-9337-4b5ee7d37bea.jpg" title=" 5.jpg" alt=" 5.jpg" / /p p 今年11月,Loxo Oncology和拜耳(Bayer)公司带来的Vitrakvi,也成为了精准疗法的最佳注脚。作为一款针对特定遗传变异,而非肿瘤病发部位的抗癌药,Vitrakvi所能治疗的NTRK基因融合实体瘤,包含了乳腺癌、结直肠癌、肺癌、甲状腺癌等癌症种类。这一新药的批准,也是癌症疗法从“基于癌症在体内的起源”转向“基于肿瘤的遗传特征”这一演变过程中的重要里程碑。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/a78f8fd3-b2f8-44fe-ac1e-31f32f7eff39.jpg" title=" 6.jpg" alt=" 6.jpg" / /p p 同样是在今年,我们迎来了能够预防偏头痛,且拥有创新机制的新药——CGRP抑制剂。来自安进的Aimovig,Teva的Ajovy,以及礼来的Emgality,为全世界受偏头痛困扰的诸多患者带来了全新的控制方案。 /p p 此外,一些新颖的疗法也引起了业内的热议。由Fresenius Kabi USA带来的Omegaven是一款鱼油成分,而GW Research带来的Epidiolex则是第一款获批的大麻活性成分。 /p p 我们也很高兴地看到,多家药明康德集团合作伙伴的新药在今年获批:中裕新药的Trogarzo是FDA十年来批准的首款创新HIV疗法,也是首例在中国生产、经美国FDA批准进入美国市场的无菌生物制品 Agios带来的Tibsovo是史上第一款获批的IDH1抑制剂,可治疗带有特定突变的急性骨髓性白血病患者 而Amicus Therapeutics的Galafold也是首款治疗成人法布里病的创新口服疗法。 /p p strong 后记 /strong /p p 从新药的获批数量与质量看,2018年是不凡的一年。但需要指出的是,今年中枢神经系统领域获批的新药数寥寥无几,依旧是行业需要解决的研发挑战 免疫疗法组合也依然有待更多的开发 此外,考虑2017年递交的新药申请数高达57,而2018年目前的数据仅为2017年的四分之叁,2019年的新药获批数,有可能会出现下滑。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/c2668f36-65aa-4197-804d-08dd22895d2a.jpg" title=" 7.jpg" alt=" 7.jpg" / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " ▲新药申请数在2018年出现了一定的下滑(图片来源:FDA) /span /p p 但我们相信,新药研发的步伐不会因此放缓。今年,美国FDA对监管框架进行了多次调整,“无缝临床试验”的框架已经浮出水面,阿兹海默病与非酒精性脂肪性肝炎等难治疾病有望采纳新的临床终点,而基因疗法,微生物疗法,人工智能与数字疗法的监管也逐渐清晰。 /p p 我们期待在创新的监管框架下,更多创新医药产品能够问世,为全世界的患者带来福音! /p p span style=" color: rgb(127, 127, 127) " strong 附:2018年FDA新药完整列表(药明康德整理,点击图片可查看大图) /strong /span /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201812/uepic/36dc84f3-1535-4aeb-8278-bd401927160b.jpg" style=" " title=" 8.jpg" / /p p style=" text-align: center " img src=" https://img1.17img.cn/17img/images/201812/uepic/f36ef1d3-5b18-4b2c-8cc3-0009a6c31a31.jpg" style=" " title=" 9.jpg" / /p



















 我要推广仪器
我要推广仪器
 下载APP
下载APP