大家好,我想问一下大家在试验中有没有遇见这样的问题:我们用的仪器型号是Beckman P/ACE MDQ,用紫外检测器,所用样品需要的检测波长是214nm.在进样完成后出现 "UV Autozero failed, check lamp or filter wheel position" 首先我想说我试过200,254nm滤过片,这些正常,能在进样完成后进行下一步的分离。这说明不是毛细管内部及窗口的问题。 其次214nm的滤光片没有发霉 新的学期刚开始,一个假期仪器没用,刚用就出现这样的问题。希望路过的各位知道的帮一下忙,我在这先谢大家了。
求助:用氢氧化钠中和后的乙酸钙为何呈现黄色?在测定土中阳离子交换量时,配制好0.5mol/l的乙酸钙是无色,移取50ml,加入酞指示剂,用0.02mol/l氢氧化钠滴定至微红色(极浅的红色),计算出应该加入到乙酸钙中2mol/l氢氧化钠的量,但经常是还没有加够氢氧化钠的时候,就出现了黄色。加完后是比较深的黄色,显然这个颜色极不正常。这种状况已经出现多次。想问一下大家,这种情况是乙酸钙的质量问题吗?你们又是用的哪个厂家的乙酸钙呢?
在做色牢度评级时发现粘贴后的样品和未粘贴的样品评定出来的级别是有差别的,一般标准要求先评级后贴样,对于这种情况容易导致样品混乱,不知道其他实验室有多少是先贴样后评级的?
版面出现的信息:今日排行:20 今日发帖:17 本版主题:6330 本月发帖:427打开帖子后出现的信息:今日排行:30 今日发帖:12 本版主题:6329 本月发帖:422它们的意义不一样?分别代表什么?
什么条件都和前一天一样,但是今天进样后发现多了一个杂质峰,今天进了两针纯乙腈,纯乙腈就有峰,然后后面进的所有处理后的生物样本在相同位置都有峰,请问是怎么回事啊
老化后进样出现这种峰,好像没有峰一样(响应值很低),调谐报告正常,请问有什么原因要如何解决[img]http://ng1.17img.cn/bbsfiles/images/2018/01/201801250902105449_4217_3191600_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/01/201801250902107729_5514_3191600_3.jpeg[/img]
我用的是毛细柱SE-30,昨天老化了柱子,老化温度程升至290度,老化后基线稳定了,但是不进样按照我分析的程升温度空白走基线时发现在260,270,280度出现了杂峰,我一共走了三次,并且每次杂峰出现的时间都不一样,请教一下这个情况是不是柱子没有老化好呀。
请大家分享下自动进样器出现故障的时候怎么处理?最近发现所用的7683B自动进样器在中途进样时出现故障,但进样针不涩,把针取下来后运行空针也是正常,但按装针后就会闪红灯。
液相色谱平衡柱子时基线很平稳,一进样后就出现大量毛刺。更换色谱柱,新配样品,清洗管路,进样口,检测器后都没有改善,请问这是什么问题呢?(已经出现好多天了,以前都是正常的,忽然就变这样了)[img=,400,224]http://ng1.17img.cn/bbsfiles/images/2018/05/201805081323183509_9165_3406574_3.jpg!w690x388.jpg[/img]
我今天进样之后发现样品基线一直上飘,之后相应降低了氢气和氧气的压力表,再进样发现就正常了,我想知道为什么降低一点就正常了
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]老化柱子后进样第一针出现包峰,后面的正常,第一张是空白,第二张系统适用性,第三张是样品(进样之前老化了柱子),第四张是样品(和第三张是一个进样瓶)[img]http://ng1.17img.cn/bbsfiles/images/2018/07/201807211115268641_33_3438808_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/07/201807211115272381_6208_3438808_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/07/201807211115274701_6475_3438808_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/07/201807211115271674_2661_3438808_3.jpeg[/img]
瑞利WFX-210富氧是怎样使用的,是先烧乙炔,然后加氧气入去一起烧吗,操作安全吗?
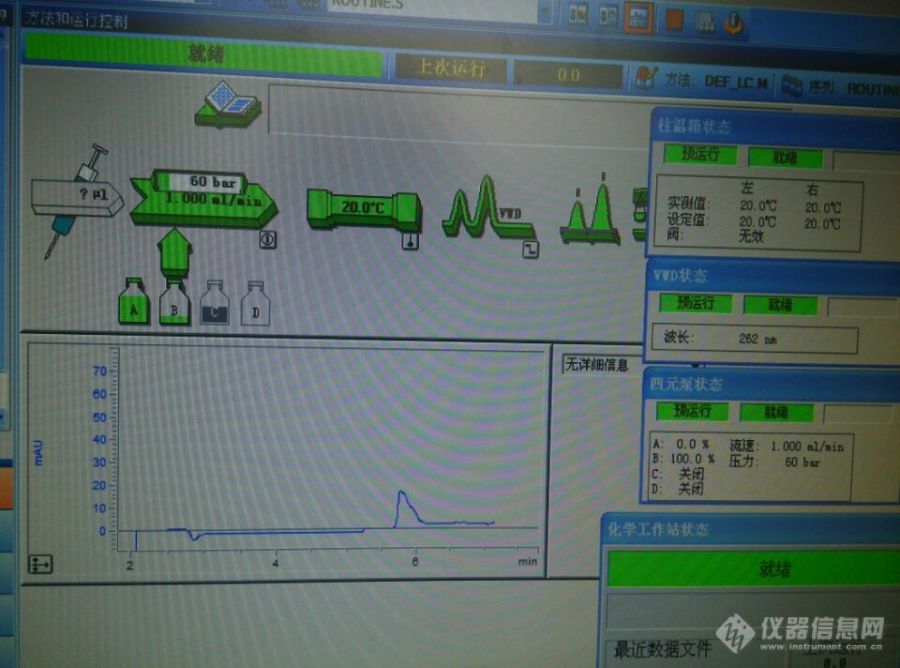
今天用Agilent 1200型高效液相色谱仪测样时,样品测完,停止时间过了之后,然后曲线就一直往上走,一直都没有停的趋势(出现很大的峰),之后更改流动相的比例(流动相大概占80左右)曲线又走平了,今天两次进样都是这种状况,不知道怎么回事,在机器关闭之前用100的甲醇流动相走20分钟,基线平了,之后关闭机器的时候一关泵就会出现曲线一直往下走的现象,走甲醇的时候也会出现如下图的现象,不知道怎么回事,本人是新手,请各位高手们指点一下,谢谢各位[img=,690,512]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021313002080_3708_1773263_3.jpg!w690x512.jpg[/img]
为什么只要我一进样,电流就会往下降?所有的峰都并在一起,完全分不开等峰出完后,电流就又恢复成一条直线了我试了好几种缓冲液,结果都是一样的如果不进样,用缓冲液跑空针的话,电流是正常的一条直线我要分离的物质主要是生物碱,木脂素等成分用乙酸乙酯提取后,再用甲醇溶的,进样前都已经超声,过滤请问出现这种情况,是我的样品制备方面的问题吗?有知道的朋友请下忙,谢谢了!!
气相保留时间延后是什么原因?与原来同样条件同一柱子,进样后保留时间滞后且出现原来不存在的峰,而且每进一针保留时间就后延一点,现在既无法定性也无法定量,请高手不吝赐教。急!!!三路气源都检查了,不漏;进样垫和衬管都换新的了;检测器喷嘴也清洗了。我束手无策,急待援兵啊!续更换另一根柱子还是如此!柱头也切了,于事无补啊。郁闷再续样品中出现的杂质峰,每进一针,峰就变大,老化柱子好些,但仍存在,而且继续进样,杂质峰又变大了。什么原因?
水氧气捕集井一般能用几瓶气?(工程师说一般2到3瓶),而我们是综合捕集井,没有指示,不知道什么时候失效,应该更换。想和大家讨论一下:捕集井失效之后会出现什么状况?会不会不进样时,基线上出现空气峰等不明物质???
进样后不出峰1、 无载气2、 进样器漏或堵3、 色谱柱链接处严重漏气4、 火焰熄灭5、 没有极化电压6、 信号线断路7、 汽化室或柱室温度太低8、 仪器信号值偏移太大9、 进样垫漏气10、 喷嘴漏气11、 毛细管分流太大12、 热导桥流未加13、 电子捕获检测器进样量过大14、 电子捕获检测器脉冲电压选择不对15、 色谱柱对样品严重吸附16、 热导桥流太低17、 毛细管接口处断裂
色谱图是以组分的流出时间(t)为横坐标,以检测器对各组分的电信号响应值(mV)为纵坐标得到的曲线图。色谱图上可得到一组色谱峰,每个峰代表样品中的一个(或多个)组分。当色谱峰峰尖向下,响应值为负mV时,就是负峰(或称之为倒峰)。什么原因造成进样后出现负峰?
怎么解决高效液相进样后总会出现波型峰HPLC-1200高效液谱,在进样后总有一个波型峰出来,出峰时间也比较固定,怎么把它给消灭掉?
基线基本稳定,但样品进样后出现一个个的波浪状色谱峰是怎么回事
不知为什么会出现M型峰,洗过柱子后还是一样。就连标样峰也是M型,请问是怎么回事?
GC中第一针正常,后面进样多出现几个杂质...每针中间进空白都不顶用...溶剂为DMSO,不知道大家遇到过类似的情况没有?每次进第一针都好着,后面的每次都多出几个杂质峰

进样前压力是80左右,进样后压力到120然后降到50用时大概3分钟,之后稳定在50是怎么回事?http://ng1.17img.cn/bbsfiles/images/2015/07/201507261922_557314_2947586_3.jpg
各位老师和专家,你们好。我们实验室用的仪器是[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] MS QP2010plus,色谱柱使用的是用来分析脂肪酸的sp2560极性毛细管色谱柱。我们在一次没有卸真空,但有将进样口温度降到50°以下,色谱柱30°以下和离子源温度150°以下的情况下,更换了衬管。换了衬管以后,在14 19 24 29min的位置时常出现硅氧烷峰,在38-50min的位置一直出现长链烷烃峰。有时候严重点,会出现后面两幅图的情况,空针下,硅氧烷峰消失,但是长链烷烃峰依旧出现。请问硅氧烷峰是隔垫流失或柱流失的标志吗?那长链烷烃峰又是什么原因导致?我们试过更换隔垫和衬管,问题依旧存在,请问有什么办法可以排查出什么问题,以及怎么解决吗?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/12/201912181122224053_1160_3229859_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/12/201912181122229432_2417_3229859_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/12/201912181122233502_2596_3229859_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/12/201912181122236373_1427_3229859_3.png[/img]
在做液相的时候,出现同一个样品溶解后,多次进样有杂质峰一直变大是什么原因呢?我这有一个项目是使用乙腈:甲醇做的流动相,样品是用乙腈溶解的,一个样品现配现做杂质是0.8%,走样需要20min,走完后再打刚才配制好的同一样品同一杂质的含量为4%,这是什么原因引起的,怎么改善呢?
我们用的是福立9790II色谱仪,进样后,进样标志一直是灰色的,一直不显示进样结束,也不出现基线,问下各位,可能是什么原因造成的,如果要维修的话费用大致是多少。
求助:有一台液相,空白基线没有问题。但只要一进样主峰后某个固定时间就有一个小峰。做了以下几点确认:1.更换新色谱柱,仍然有。2.同样样品换另外一台液相,没有。3.重新找出以前这台机,测试没有问题的样品测试,有。从以上几点确认,看不出哪有问题呀,说机器的问题,也说不过呀,空白都没有。
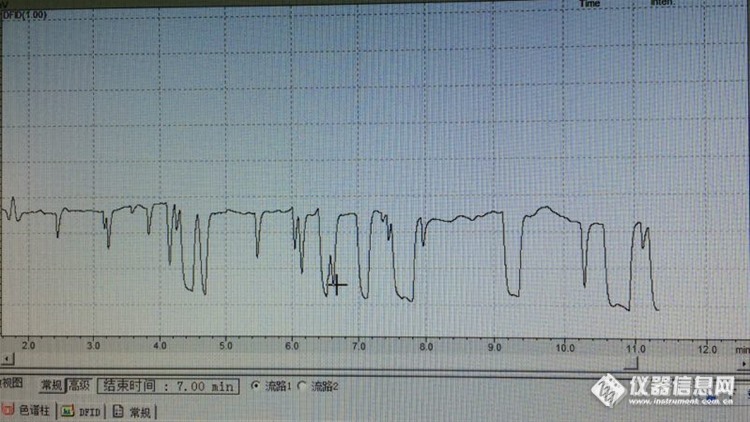
测定非甲烷总烃,本来基线正常,进一次样之后基线即出现不正常[img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/07/201707232101_01_3227746_3.jpg[/img]
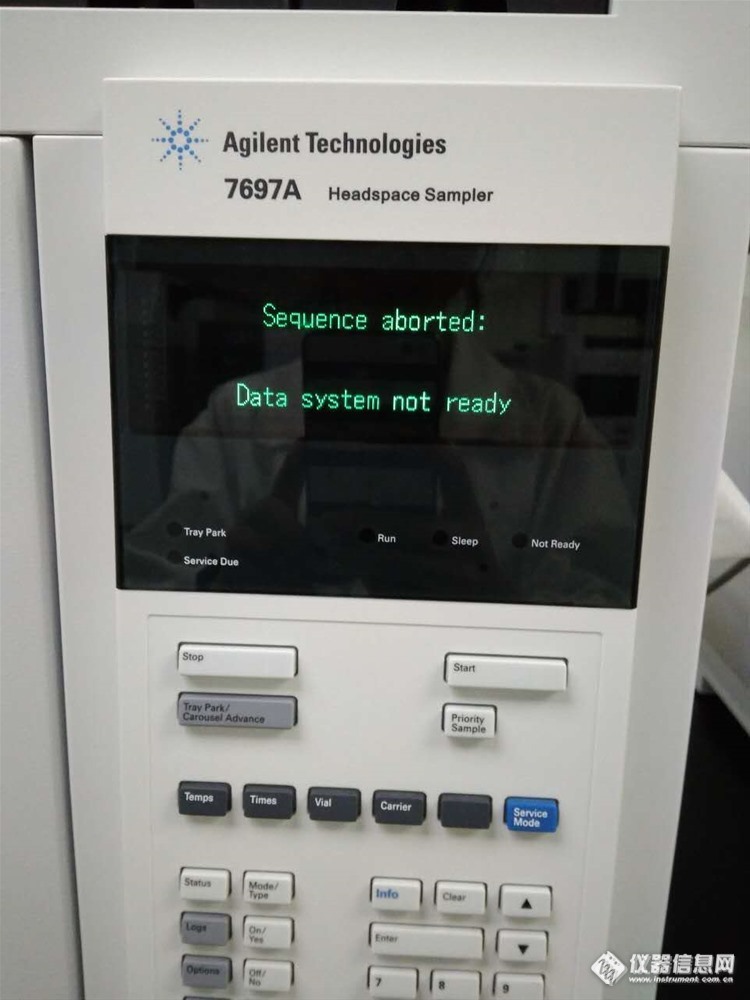
序列跑样至第六针出现“Data system not ready”.重复后结果还是一样的,应该不是针松了,[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2017/08/201708022115_01_3227746_3.jpg[/img]
最近我们实验室安捷伦7890-5975做样比较频繁,发现有样品保留时间漂移,自动调谐发现有轻微漏气,开始发现后,先更换了进样口密封垫,同时,按原来工程师指导的,把进样口和传输线端的柱螺母都上紧了些,好了一阵子,这两天自动调谐又发现有轻微漏气,关机,更换了进样口及传输线石墨垫后,无法抽真空了。MS面板显示pump dowm一会之后泵就停止了,前级泵有明显的水流的汩汩声,略微有点机油味,不知道是哪个地方的问题,请大神们帮忙指导下。十分感谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP