小妹医学出身,对仪器检测方面一窍不通,现在做博士课题,我计划检测动物体内(兔)骨骼和血液中的26Mg含量的变化,26Mg为Mg的稳定同位素,在自然界中占11.17%,24Mg占78.7%,25Mg占10.13%,秋月芙蓉告诉我ASS AES都不行,得用MC-ICP-MS检测,并建议我到这里发帖子求助,在这里表示感谢!请问怎么检测动物体内的26Mg哪?哪里可以做? 我联系了地质科学院矿产资源研究所,被拒绝了。
我现在做藻体内DDT检测,将离心后出来的藻泥用高氯酸和冰醋酸破坏后,再用正己烷萃取,然后浓硫酸磺化,20%硫酸钠溶液水洗,无水硫酸钠脱水,定容,GC-ECD检测。但是杂峰太多,基线漂的老高了,看文献用的就是这种方法,为啥杂峰这么多呢?求高手指点,谢谢
各位亲们,我现在正在做植物体内的醛类物质检测,经过查询方法,基本是用DNPH衍生后,再进行HPLC检测。但是,我现在在处理标准醛类物质,比如甲醛、丙烯醛等物质的衍生时,发现文献中的方法不正确,有谁正在做类似实验呢?只要是会处理标准醛类物质的DNPH衍生就行。不胜感激。
那位有生物体内石油烃的监测方法(荧光分光光度法),英文的,最好是标准。
鸡体内氯霉素残留的检测方法及消除规律研究[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=83715]鸡体内氯霉素残留的检测方法及消除规律研究[/url]
如何通过质谱检测的质荷比推断药物在体内代谢时结构的变化
请问有谁知道国内有哪家检测机构可以检测鱼类体内有机砷和无机砷的吗?在这里先谢过了![img]http://simg.instrument.com.cn/bbs/images/brow/em09511.gif[/img]
请问:动物体内丙烯腈(ACN)代谢,脏器中ACN气象检测有人做过么?查到的相关资料都是用同位素标记法检测的!
[font=宋体][size=10.5000pt][font=宋体]荧光法温度检测技术能否应用到植物体内的细胞逆境应答温度监测[/font]?[/size][/font]
农药被农作物吸收,在植物体内代谢,有什么好的方法对代谢物进行定性分析呢?请各位大侠指教!
请问我建好了一种牛兽药残留检测方法,想做体内验证,可以5.6只牛注射后一段时间后采样检测吗?并不测这个方法在牛体内的残留消除规律,只是验证方法的可行性
体内药物分析方法的设计与评价 一、分析方法的设定依据:(一)重视并做好文献总结、整理工作;(二)充分了解待测药物的特性及体内存在状况;(三)明确测定的目的要求;(四)实验室条件。二、方法建立的一般实验步骤:(一)以纯品进行测定:以一定量纯品按拟定方法进行测定。求得浓度与测定响应值之间(如吸收度、色谱峰高或面积等)的关系,浓度线性范围,最适测定浓度,检测灵敏度,测定的最适条件(pH、温度、反应时间)等等。(二)以经过纯化处理过的空白样品进行测定;(三)空白样品添加标准后的测定:血样等样品中添加一定量标准品后进行测定,求得样品回收率数据,检验生物样品对测定有无干扰等。(四) 体内实际样品测定:有时用体外建立的方法去测定体内取得的实样时,会得出错误的结论。故要强调对药物体内过程有一定程度的了解。有时也采用专属性强、已证明适用于体内实样测定的步骤和方法作为对照测定,并以此来检验所建立的方法的实际可行性。三、方法的评价:(一)准确度(Accuracy):测定结果与真值的符合程度。常用回收率(Recovery)数值间接反映测定的准确程度;也可通过与其他已建立的方法进行比较的办法(参比方法)来加以反映。回收率100%当然好,但很难达到。重要的是每次测定要保持稳定。(二)精密度(Precision):测定结果与平均值的偏离程度。测定间偏差越小,对测定的要求也越高(花费大);浓度与RSD值间存在反比关系,RSD在10%以内的方法可认为是可接受的。(三)灵敏度(Sensitivity):“一种方法可以检测出有关化合物的最小量”。常用最低检测限(Limit of detection,LOD)或最低检测量(Limit of quantification;LOQ)来表示。LOD范围在ng(10-9g)~10-18g。(四)专属性或选择性(Specificity or Selectivity):是指测定的信号(响应)是属于被测药物所特有的。若有干扰就需改进测定方法或改用具有分离能力(如色谱法的专属性较吸收光度法为高)的方法或专属性较强的方法进行。(五)不同方法测得结果的相关程度(Degree of correlation)的比较:用一有相当专属性和可靠性的方法与新建方法同量测定,以相关系数γ(Correlation coefficient)表示相关程度。γ一般要求在0。95以上。此外,还应从方法的可靠性、每个样品测定耗时多少、操作的难易及技术要求及仪器、设备要求、费用多少等等方面加以考虑。
如果你体内的重金属超标,可能会有头疼、胃口不好、浑身乏力的症状,但均不明显,有可能与其它的病症混淆。而毛发检验是目前最有效的检测人体内各项微量元素含量的方法,只要检验0.2克的头发,就可以知道体内26种微量元素(包括6种有害元素)的均衡状况。人体血液中的微量元素通过与蛋白质结合的形式,被排泄积蓄于毛发中.毛发反映了人体内微量元素的长期积累状况。 而血液检查和尿液检查反映的是排泄速度较快的短期状况。另外,由于体内平衡的缘故,血液检查有时很难准确反映微量元素含量。

[img]http://ng1.17img.cn/bbsfiles/images/2008/05/200805121951_89037_1626751_3.jpg[/img]这是2005年1月18日拍摄的生活在南极洲阿德利岛上的企鹅。美国研究人员5月9日说,在全球大部分国家禁用杀虫剂DDT数十年后,南极阿德利企鹅体内仍检测出这种有毒物质,且含量多年来始终不降。他们认为,这是因为DDT被“储存”在冰川中,持续影响南极生态环境。(新华社/路透) 美国研究人员5月9日说,在全球大部分国家禁用杀虫剂DDT数十年后,南极阿德利企鹅体内仍检测出这种有毒物质,且含量多年来始终不降。 他们认为,这是因为DDT被“储存”在冰川中,持续影响南极生态环境。 美国弗吉尼亚州海洋科学研究所的海鸟研究专家海迪盖兹说,研究人员早在20世纪60年代初就从阿德利企鹅的脂肪内检测出DDT。但令研究人员感到惊奇的是,尽管这种有毒农药已在大多数国家被禁用数十年,它在企鹅体内的含量却一直没有下降。 盖兹解释说,这可能是因为DDT等化学物质被蒸发后经大气层传播到南极,然后冷凝,“储存”到冰川中。证据之一是研究人员在冰川融化后的水中检测出DDT。 尽管南极企鹅体内的DDT含量不降,但盖兹领导的这项研究发现,在过去数十年内,北极野生动物体内的DDT含量大幅下降。盖兹说,目前在企鹅体内检测出的DDT含量还不足以伤害它们。 这项研究结果发表在美国《环境科学与技术》(Environmental Science & Technology)月刊上。[em0812] [em0815] 中国心
体内药物分析 :是通过分析的手段了解药物在体内(包括实验动物等机体)数量与质量的变化,获得各种药物代谢动力学的各种参数和转变、代谢的方式、途径等信息。从而有助于药物生产、实验、研究、临床等各个方面对所研究的药物作出估计与评价,以及对药物的改进和发展作出贡献。 体内药物分析任务和对象的特点: 1、被测定的药物和代谢物的浓度或活性极低;2、样品中存在各种直接或间接影响测定结果的物质,大多需要分离和净化;3、样品量少,尤其是连续测定时,很难再度获得完全相同的样品;4、工作量较大,随着工作的深入开展,会成倍地甚或按指数级数增加;5、往往要求很快地提供结果,尤其在毒物检测工作中;6、实验室应有多种检测手段,可进行多项分析工作;7、测定数据的处理和阐明有时不太容易。 样品的种类、采集和储存一、样品的种类和选取原则:(一)血样:血浆(plasma)和血清(serum)是体内药物分析最常采用的样本,其中选用最多的是血浆。因血浆中的药浓可反映药物在体内(靶器管)的状况。而且血浆中药物浓度的数据报道较多,可供借鉴。血浆是全血(whole blood)在加肝素、枸橼酸、草酸盐等抗凝剂的全血经离心后分取,量约为全血的一半。血清则是在血液中纤维蛋白元等影响下,引起析出血块,离心取得。血块凝结时往往易造成药物吸附损失。全血也应加入抗凝剂混匀,以防凝血。对大多数药物来说血浆浓度与红细胞中的浓度成正比,所以测定全血也不能提供更多的数据,而全血的净化较血浆与血清麻烦,尤其是溶血后,血色素等可能会给测定带来影响。但是一些可与红血球结合或药物在血浆和血球的分配比率因不同病人而异的情况下,则宜采用全血。血样采取量会受到一定的限制,血样取样时间间隔问题也常随测定目的不同而异。目前大都是测定原型药物总量。当药物与血清蛋白结合率稳定时,血药总浓度可以有效表示游离药物的浓度。但对低蛋白症或尿毒症患者,药物结合率降低,则在通常安全有效的血药总浓度中,游离型药物浓度可显著增加。(二)尿样(urine):尿样测定主要用于药物剂量回收研究、药物肾清除率和生物利用度等研究,以及测定代谢物类型等。体内药物清除主要是通过尿液排出,药物可以原型(母体药物)或代谢物及其缀合物形式排出。尿液药物浓度较高,收集量可以很大,但尿液浓度通常变化较大,所以宜测定一定时间内尿中药物的总量(如8、12、24小时内的累计量),需记录排出尿液体积及尿药浓度。尿药浓度改变不直接反映血药浓度,受试者肾功能将影响药物的排泄。尿中药物大多呈缀合状态,测定前要将缀合的药物游离。此外,采集尿液不可能在较短时间内多次取样,排尿时间较难掌握(尤其是婴儿),同时也具有不易采集完全的缺点。(三)唾液(saliva):唾液中的药物浓度通常与血浆浓度相关。样品易得,取样无损害,尤易为儿童接收。有些可从药物唾液浓度推定血浆中游离药物浓度。但有些蛋白结合率较高的药物在唾液中的浓度比血浆浓度低得多,需高灵敏度的方法才能检测。唾液pH值6.9±0.5,每日分泌量1~1.5L,含有的主要电解质有Na+、K+、Cl-、HCO3 -等,主要有机成分是粘液质和淀粉酶。采样一般是在漱口后15分钟,收集口内自然流出或经舌在口内搅动后流出的混合唾液(吸管内吸附的少量唾液用稀释液洗出),用2000~3000rpm离心15分钟,小心吸取上清液,进一步分离、净化。也可采用物理(嚼石蜡片、小块聚四氟乙烯或玻璃大理石)或化学(酒石酸、维生素C)的方法刺激,在短时间内可得到大量唾液,但药浓也可能会受到影响。(四)其它:乳汁、动物脏器组织匀浆等。二、样品储存和稳定性考察:取样后最好立即进行分析,冷藏(4℃)、冰冻(-20℃)有时也不能完全保证样品不起变化。尿液是很好的细菌生长液,若需收集24小时或更长时间的样品或不能立即测定的,应置冰箱冷藏或加防腐剂(1%甲苯、过饱和氯仿)保存。分析样品贮存时应考虑:储存条件;样品在贮存中会对分析结果产生什么影响;评述样品稳定性时会发生什么问题;如何预防或校正不稳定样品的分析结果。 测定前样品的制备 除少数体液经简单处理后直接测定外,通常在最后一步测定前要采取适当的样品制备,即进行分离、净化、浓集、必要时尚需对待测组分进行化学改性,为测定创造良好条件。一、样品的制备要考虑:药物的理化性质、待测物的浓度范围、药物测定的目的、选用的生物体液和组织的类型、样品制备与分析技术的关系。二、蛋白质的处理:是测定血浆、血清、全血及组织匀浆等样品中药物时的最先处理步骤。(一)加入沉淀剂和变性试剂:硫酸铵是经典的蛋白质沉淀剂,它与蛋白质分子竞争系统中水分子,而使蛋白质析出。阴离子型沉淀剂(三氯醋酸、高氯酸、钨酸、焦磷酸)与带电荷的蛋白质在氏于等电点的pH时形成不溶性盐;反之,阳离子型沉淀剂(含锌盐、铜盐)与蛋白质分子中带阴电荷的羧基,在高于蛋白质等电点时,形成不溶性盐。有关机制不十分清楚。(二)加入可与水混合的有机溶剂:乙醇过量存在时,能使与蛋白结合状态的药物释放可将混合物离心,取上清液(含药),但这不能解决样品的净化问题。蛋白沉淀法对于与蛋白结合力强的药物的回收率较差。也有采用酸消化法(Acid digestion)使药物自蛋白结合处释出,但常导致药物的分解。
请问我要测定昆虫体内的5-羟色胺的含量,请问采用哪种色谱仪器检测好呢?还有采用什么方法提取比较好呢,请各位高手赐教,非常感谢
[img=小动物体内荧光成像系统]http://www.f-lab.cn/Upload/FluorVivo-system.jpg[/img][b][url=http://www.f-lab.cn/vivo-imaging/fluorvivo.html]小动物体内荧光成像系统fluorvivo[/url]应用[/b]表达荧光标记的小动物荧光筛选;肿瘤转移负担评价;药效试验内化物质的药代动力学;荧光物质的定量测量,如肿瘤负荷;连续或时间推移监测。小动物体内荧光成像系统:[url]http://www.f-lab.cn/vivo-imaging/fluorvivo.html[/url]
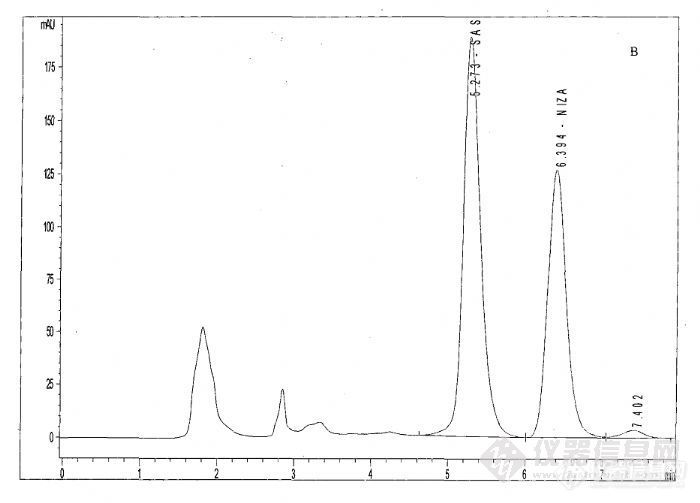
【作者中文名】张玲;【导师】王婷;【学位授予单位】兰州大学;【中文摘要】目的:建立高效液相色谱法测定人血浆和尿样中尼扎替丁浓度的方法。研究健康受试者单次和多次静脉滴注尼扎替丁注射液(100mg)后的血药和尿药药代动力学特征,计算其在健康人体内的药代动力学参数,描述其在体内的分布、代谢、排泄规律,并对该剂量静脉滴注后的安全性进行初步评价,为制定Ⅱ期临床试验方案及临床用药提供依据。 方法:本试验采用平行设计,筛选10名健康志愿受试者(男女各半),按体重随机排序,单次(100mg)和多次(100mg/次,3次/日,连续用药6天)静脉滴注尼扎替丁后,采集血样和尿样。色谱柱为Diamonsil C18 (150×4.6mm,5μm);流动相:V:V=L腈:0.05mol/LK2HPO4(含三乙胺1.0%v/v,用85%磷酸调pH至6.5)=17:83,流速为0.9mL/min;柱温:40℃;DAD检测器,检测波长320nm。采用RPHPLC-UV法,以水杨酸为内标测定尼扎替丁血药及尿药浓度,DAS软件计算药动学参数。 结果:1、尼扎替丁血药浓度在0.0117-6μg/mL范围内具良好线性关系(r=0.9999,权重W=1/C),方法回收率在96.2%~105.2%之间,提取回收...http://ng1.17img.cn/bbsfiles/images/2012/08/201208131811_383608_2379123_3.jpg
8月25日,国际环保组织绿色和平给记者发来的新闻稿中称,在对取自长江的两种常见食用鱼进行检测后,发现了可以[color=#0021b0]干扰内分泌并影响性发育水平[/color]的有毒有害物质,而中国尚未将这些化学物质纳入监管体系。绿色和平呼吁中国立即加强有毒有害物质监管,以减少、限制和最终消除有毒有害化学物质的使用和排放。 在最新水污染调查报告《“毒”隐于江——长江鱼体内有毒有害物质调查》发布会上,绿色和平水污染防治项目主任武毅秀说:“在取自长江上、中、下游不同城市的鲤鱼和鲶鱼体内,我们测出了被称为[color=#0021b0]‘环境激素’的壬基酚(NP)和辛基酚(OP)。[/color]这两种物质可[color=#0021b0]导致雌性性早熟以及雄性精子质量下降、数量减少等性发育和生殖系统问题[/color]。” 除了壬基酚和辛基酚之外,绿色和平在这些来自重庆、武汉、马鞍山和南京的野生鲤鱼与鲶鱼体内还检测出了广受国际关注的持久性有机污染物[color=#0021b0]全氟辛烷磺酸(PFOS)[/color],部分鱼体内还检测出了[color=#0021b0]汞、铅和镉等重金属[/color]。这些化学品被大量的用于工业生产之中。壬基酚和辛基酚是洗涤剂、纺织产品、以及皮革涂饰中极为常见化学原料,而全氟辛烷磺酸则被广泛应用于纺织品、地毯、造纸、防水涂料、消防泡沫等产品中。 长江是中国第一大河,养育了中国近三分之一的人口。近10年来,长江流域的工业污水排放量迅速增长了67%。大量的有毒有害物质不仅给长江流域本已脆弱的生态系统带来了更大的压力,也为我国居民健康和生态环境带来巨大威胁。 绿色和平表示,壬基酚已经在我国很多的河水及沉积物中检出,包括重庆地区的饮用水。一份最近的研究也显示,中国婴幼儿和儿童的血液样本中发现了全氟辛烷磺酸的存在。 “这些有毒有害物质在[color=#0021b0]生物体内具有累积性[/color],因而可以通过食物链进入人体,形成健康隐患。”武毅秀说:“由于这些有毒有害物质对环境和健康巨大的负面影响,许多发达国家和地区已经将其列为禁止或限制使用的化学物质,因而其产量在这些国家已大幅减少。” 然而由于中国尚未将这些化学物质纳入监管体系,其在中国的产量却在迅速增加。壬基酚(NP)在中国的产量自1995年至2003年增长了一倍以上,占到世界年产的26%,而全氟辛烷磺酸(PFOS)的产量从2004年到2006年短短三年里增长了四倍。 武毅秀说:“更高的产量势必会增加人和其他生物接触到这些化学物质的风险,中国公众正毫无保护地暴露于不断增长的有毒有害物质威胁之下。” 根据绿色和平提供的资料显示,针对这些有毒有害物质的全球限制和淘汰行动正在进行中。其中最重要的行动是《关于持久性有机污染物的斯德哥尔摩公约》在去年五月的缔约方大会上同意将PFOS等九种持久性有机污染物列为公约受控污染物名单,从而使得PFOS和臭名昭著的DDT等物质一起,成为全球限制和淘汰的目标。该修正案将与明天,也就是2010年8月26日正式生效。 除此之外,欧盟早在2001年的水框架指令(EuropeanUnionWaterFrameworkDirective)中,将壬基酚列为“优先有害物质”,这意味着在2020年之前,欧盟成员国将最终完全停止向环境中排放壬基酚。而欧盟在2006年也通过了限制PFOS生产与销售的指令。 美国早在2000年就宣布禁用PFOS,直接推动了3M公司这一PFOS主要生产商宣布在2003年之前完全停止其生产。而就在过去一周,美国环保署出台了针对NP的行动计划。除了支持洗涤剂行业淘汰NP的行动之外,环保署还宣布将立法要求企业按照有毒物质排放清单(TRI)的规定汇报其使用和排放情况,并有可能彻底禁止NP。 中国政府正在大力治理汞、铅和镉等重金属引发的污染问题,但对于本次检出的全氟辛烷磺酸、壬基酚和辛基酚等有毒有害物质的监管,仍处于起步阶段。“有毒有害物质污染正在成为中国水污染的新课题。”武毅秀最后补充道:“中国政府的当务之急是尽快摸清当前使用和排放的有毒有害物质的情况,并加速出台管理规定,来逐步减少、限制并最终消除有毒有害化学物质的使用和排放。”
北京大学治疗药物监测与毒理中心(北京大学第三医院药学实验室)主要从事临床药理及治疗药物监测工作,进行体内药物及小分子生物标志物的检测方法的开发,有丰富的生物分析方法开发经验。先后获得了科技部“十一五”及“十二五”重大专项国际标准化药物临床试验研究技术平台建设——I期实验室建设项目的支持,同时获得了卫生部重点专科建设项目的支持。硬件方面,目前实验室拥有液质联用仪4台(ABSciex 5500QTrap、ABSciex 4000、API 3000、Agilent 6410)、ICP/MS 一台、液相色谱仪4台。软件方面,实验室于2009年首批通过了ISO/IEC检测与校准实验室认可,有完善的质量管理体系。国际合作方面,本实验室近年来与阿斯利康公司和美迪恩斯株式会社合作,质量管理方面得到了进一步提升,管理水平与国际接轨。 现招收进修人员若干名,时间半年以上。要求如下:1、药学或药物分析相关专业本科以上学历;2、有液相或液质相关经验者优先。http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif3、责任心强,踏实认真。联系人:张现化电话:133 9153 9521邮箱:zhangxianhua432@163.com北京大学治疗药物监测与毒理中心北京大学第三医院药学实验室
体内药物分析是通过分析的手段了解药物在体内(包括实验动物等机体)数量与质量的变化,获得各种药物代谢动力学的各种参数和转变、代谢的方式、途径等信息。从而有助于药物生产、实验、研究、临床等各个方面对所研究的药物作出估计与评价,以及对药物的改进和发展作出贡献。 体内药物分析任务和对象的特点:1、被测定的药物和代谢物的浓度或活性极低;2、样品中存在各种直接或间接影响测定结果的物质,大多需要分离和净化;3、样品量少,尤其是连续测定时,很难再度获得完全相同的样品;4、工作量较大,随着工作的深入开展,会成倍地甚或按指数级数增加;5、往往要求很快地提供结果,尤其在毒物检测工作中;6、实验室应有多种检测手段,可进行多项分析工作;7、测定数据的处理和阐明有时不太容易。样品的种类、采集和储存一、样品的种类和选取原则:(一)血样:血浆(plasma)和血清(serum)是体内药物分析最常采用的样本,其中选用最多的是血浆。因血浆中的药浓可反映药物在体内(靶器管)的状况。而且血浆中药物浓度的数据报道较多,可供借鉴。血浆是全血(whole blood)在加肝素、枸橼酸、草酸盐等抗凝剂的全血经离心后分取,量约为全血的一半。血清则是在血液中纤维蛋白元等影响下,引起析出血块,离心取得。血块凝结时往往易造成药物吸附损失。全血也应加入抗凝剂混匀,以防凝血。对大多数药物来说血浆浓度与红细胞中的浓度成正比,所以测定全血也不能提供更多的数据,而全血的净化较血浆与血清麻烦,尤其是溶血后,血色素等可能会给测定带来影响。但是一些可与红血球结合或药物在血浆和血球的分配比率因不同病人而异的情况下,则宜采用全血。血样采取量会受到一定的限制,血样取样时间间隔问题也常随测定目的不同而异。目前大都是测定原型药物总量。当药物与血清蛋白结合率稳定时,血药总浓度可以有效表示游离药物的浓度。但对低蛋白症或尿毒症患者,药物结合率降低,则在通常安全有效的血药总浓度中,游离型药物浓度可显著增加。(二)尿样(urine):尿样测定主要用于药物剂量回收研究、药物肾清除率和生物利用度等研究,以及测定代谢物类型等。体内药物清除主要是通过尿液排出,药物可以原型(母体药物)或代谢物及其缀合物形式排出。尿液药物浓度较高,收集量可以很大,但尿液浓度通常变化较大,所以宜测定一定时间内尿中药物的总量(如8、12、24小时内的累计量),需记录排出尿液体积及尿药浓度。尿药浓度改变不直接反映血药浓度,受试者肾功能将影响药物的排泄。尿中药物大多呈缀合状态,测定前要将缀合的药物游离。此外,采集尿液不可能在较短时间内多次取样,排尿时间较难掌握(尤其是婴儿),同时也具有不易采集完全的缺点。(三)唾液(saliva):唾液中的药物浓度通常与血浆浓度相关。样品易得,取样无损害,尤易为儿童接收。有些可从药物唾液浓度推定血浆中游离药物浓度。但有些蛋白结合率较高的药物在唾液中的浓度比血浆浓度低得多,需高灵敏度的方法才能检测。唾液pH值6.9±0.5,每日分泌量1~1.5L,含有的主要电解质有Na+、K+、Cl-、HCO3-等,主要有机成分是粘[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]和淀粉酶。采样一般是在漱口后15分钟,收集口内自然流出或经舌在口内搅动后流出的混合唾液(吸管内吸附的少量唾液用稀释液洗出),用2000~3000rpm离心15分钟,小心吸取上清液,进一步分离、净化。也可采用物理(嚼石蜡片、小块聚四氟乙烯或玻璃大理石)或化学(酒石酸、维生素C)的方法刺激,在短时间内可得到大量唾液,但药浓也可能会受到影响。(四)其它:乳汁、动物脏器组织匀浆等。
哪位用ICP-AES测过人体内的微量元素,我想知道具体的前处理和标准溶液的浓度这两个方面的内容.多谢了.

作者:http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif宋笑丹 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif刘红梅 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif魏华 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif董迪 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif吴琳华 Author:SONG Xiao-dan LIU Hong-mei WEI Hua DONG Di WU Lin-hua 作者单位:哈尔滨医科大学附属第二医院药学部,黑龙江省高校重点实验室,哈尔滨,摘要: 目的 考察β-榄香烯脂质体在大鼠体内的组织分布.方法 建立了大鼠体内β-榄香烯测定的HPLC.色谱条件为:Diamonsil C18色谱柱(4.6 mm×250 mm,5μm);流动相为甲醇-乙腈-水(40:57:3);检测波长:210 nm;柱温:30℃.并测定大鼠尾静脉注射β-榄香烯脂质体和榄香烯注射液后血浆及组织中的药物浓度.结果 此色谱条件下血浆与组织的标准曲线、精密度等实验结果表明,该方法适于分析生物样品中β-榄香烯含量.与榄香烯注射液相比,β-榄香烯脂质体在大鼠体内的分布特性有不同程度的改变,其中β-榄香烯脂质体在肝、脾、肾组织中分布相对较多.结论 β-榄香烯脂质体及榄香烯注射液在大鼠的心、脾、肾组织中分布具有显著性差异http://ng1.17img.cn/bbsfiles/images/2012/08/201208271803_386611_2379123_3.jpg
http://ng1.17img.cn/bbsfiles/images/2010/12/201012161703_267458_1759541_3.gif 我们人类处于食物链的最高级,动物所食用和使用的饲料和药品都会通过食物链进入我们的体内,而动物中抗生素的滥用不容忽视,我们不会天天生病,没有天天吃药,可是我们却每天都在从食物中摄入抗生素等药物!!http://ng1.17img.cn/bbsfiles/images/2010/12/201012161703_267459_1759541_3.gif 动物性食品是抗生素的重要来源,兽残检测的很大一块工作也是在于此,从事这方面分析的版友们,大家都来讨论一下:自己检测的样品到底有多少此类残留,到底抗生素的使用情况有多么的严重吧!
核兹共振(NMR)在体内药物分析中,可用于药物及其代谢物的结构鉴定、代谢途径归属、定量分析以及药物与内源性物质相互作用的研究等。与其它分析方法相比,具有如下优点: ①简便性:无需对样品进行繁杂的提取或衍生化, 减少了由此带来的误差; ②无损伤性:对取样量有限的生物样品经NMR分析后还可用于其它处理, 甚至可对生物整体进行无损伤分析; ③连续性:NMR可对整体生物系统进行动态监测而不扰乱生物体内的各种平衡, 实现药物的在体分析; ④高分辨性:NMR谱线为Hz量级,能提供分子水平的结构信息; ⑤多目标性:无需进行分析条件摸索,可在同一物理条件下检测药物及其多种代谢物。 1H-NMR 已广泛用于体内药物分析。已报道的有: 氨苄青霉素、布洛芬、硝苯地平、阿司匹林、美西律等的体内样品分析。Connor等用高分辨1H-NMR (400MHz) 研究了大鼠静注羟氨苄青霉素后24h内尿样中药物的代谢情况。实验用自旋回波技术, 消除内源性物质的干扰, 增强了测定的灵敏度。尿样中共振信号在0. 5~1. 7ppm 范围内的两组峰为青霉素结构噻唑环C2上的一对偕甲基信号, 分辨清晰, 测得主要代谢物为5R , 6R 和5S, 6R 青霉素与二酮哌嗪。 Murphy等用质子去偶及NOE增益19F-NMR监测接受化疗病人的肝脏中5-FU及其代谢物(FBAL)。Campbell等利用NMR无损伤特性及表面线圈技术, 测定了不同剂量抗菌素3-氟甲基青霉素V衍生物在活体SD 大鼠体内的药物浓度。将静脉注药后的麻醉鼠置于表面线圈中,用19F-NMR 测定鼠膀胱内尿样及胸内药物浓度。 Ogiso等用13C-NMR 探讨了脂肪酸对普萘洛尔透皮吸收的影响。实验结果表明: 与月桂酸酰胺及甲酯化合物相比, 月桂酸对普萘洛尔透皮吸收的增强作用显著。普萘洛尔制剂中加入月桂酸后, 血浆中普萘洛尔浓度明显提高。本文摘自冯敏,“NMR技术在体内药物分析中的应用”,药学进展,1998,22(4
各位前辈大家好: 初来乍到请多多关照!我最近想做监测鱼体内血清中类固醇激素的实验,实验室的仪器和设备都是很齐全的,我打算用HPLC-Q/TOF去做,先定性后定量。现在有一个问题难倒我了,就是标准品因为我做内源性的激素物质的监测,雄激素、雌激素和孕激素的标准品大多是哺乳动物和人的,我有3点疑惑,请懂的人帮忙指点一下:1、有卖的雌激素、雄激素和孕激素是从鱼体内提取的吗?谁知道请帮我提供一个公司的名称被 2、如果标准品都是从人或者哺乳动物体内提取的,我在用飞行时间质谱做鱼体内监测的时候能够扫描出来吗,比如说标准品是雌二醇是从人体内提取后制成标准品的,那么我监测鱼体内的内源性雌二醇能扫描出来吗,是同一种物质吗,分子结构是否相同呢?我看一些标准中检测过外源性性激素,他们所用的标准品是怎么合成的呢,我用这种标准品能够检测我鱼体内的呢?http://simg.instrument.com.cn/bbs/images/brow/em34.gif(说的有点麻烦,不知道大家看懂没) 3、用过ABSciex家的5600+的仪器的高手们,请问在没有标准品的情况下是否无法进行监测,不用标准品有没有什么软件或者技术能分析出来物质,没有标准品无法进行定量吧!问题很棘手也很多!请大家给予帮助,初来乍到积分很少,努力攒积分吧!谢谢了!
人体内的毒素,排出不彻底,将影响人的健康,对于女性朋友来,显得更加重要,世面上有很多排毒的药物,那么他们能起到排毒养颜的效果吗,那这些毒素是通过食物直接带入体内还是在体内转化的?
测脂质体内相pH用什么仪器呢
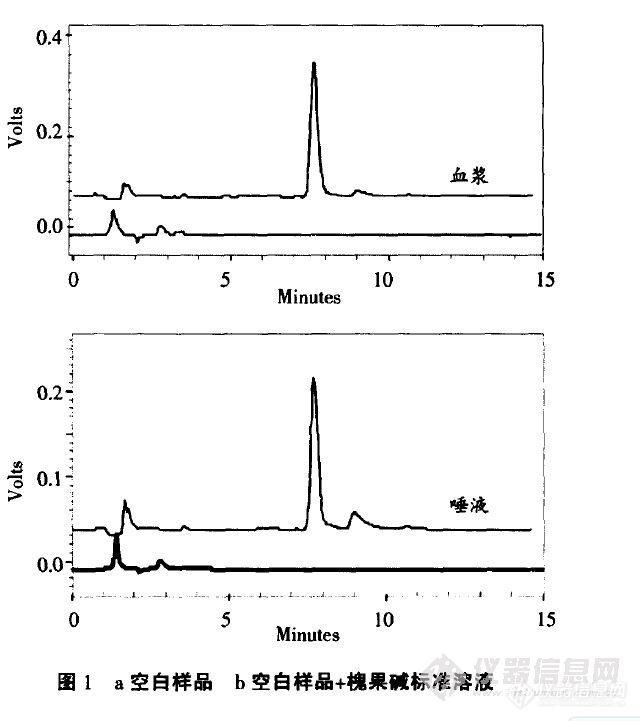
【作者】 李秋红; 张丹丹; 李纯; 韩华;【Author】 Li Qiuhong Zhang Dandan Li Chun Han Hua (Pharmaceutical School Clinical Department,Heilongjiang University of Chinese Medicine,Harbin 150040)【机构】 黑龙江中医药大学药学院临床药学教研室;【摘要】 目的:建立测定大鼠体内血浆和唾液中槐果碱含量的HPLC分析方法。方法:采用色谱柱:Diamonsil(R)C18柱150mm×4.6mm,流动相:甲醇-0.1‰三乙胺水溶液(60:40,v:v),检测波长:220nm,柱温:30℃,流速:0.8ml/min。结果:槐果碱血浆和唾液的相对回收率分别为92.7%~96.6%和94.8%~113.0%,日内、日间精密度RSD10%,血浆和唾液中槐果碱浓度在8~160μg/mL和2~40μg/mL范围内相关系数分别为0.9994和0.9995,线性关系良好。结论:首次建立槐果碱唾液样品检测方法以及用HPLC法同时测定大鼠体内血浆和唾液中的含量方法,为槐果碱的临床药动学研究提供了新的方法学基础。 更多还原【Abstract】 This is a study to determine sophocarpine in rats’ plasma and saliva using HPLC approach.We made the study under the following parameters:Diamonsil(R)C 18 Column at 150mm×4.6mm,methanol-water containing 0.1‰triethy- lamine (60:40,v:v) for mobile phase,a detection wave length at 220 nm,a temperature at 30℃,and a flow-rate of 0.8 mL/min.The study has produced the following results:a relative recovery of sophocarpine in plasma and saliva at 92.7%~96.6%,and 94.8%~113.0% respectively;the RSD of intra... 更多还原【关键词】 HPLC; 槐果碱; 血浆; 唾液; 【Key words】 HPLC; Sophocarpine; Plasma; Saliva; http://ng1.17img.cn/bbsfiles/images/2012/08/201208061030_381710_2352694_3.jpg
体内药物分析 体内药物分析是通过分析的手段了解药物在体内(包括实验动物等机体)数量与质量的变化,获得各种药物代谢动力学的各种参数和转变、代谢的方式、途径等信息。从而有助于药物生产、实验、研究、临床等各个方面对所研究的药物作出估计与评价,以及对药物的改进和发展作出贡献。 体内药物分析任务和对象的特点:1、被测定的药物和代谢物的浓度或活性极低;2、样品中存在各种直接或间接影响测定结果的物质,大多需要分离和净化;3、样品量少,尤其是连续测定时,很难再度获得完全相同的样品;4、工作量较大,随着工作的深入开展,会成倍地甚或按指数级数增加;5、往往要求很快地提供结果,尤其在毒物检测工作中;6、实验室应有多种检测手段,可进行多项分析工作;7、测定数据的处理和阐明有时不太容易。 样品的种类、采集和储存一、样品的种类和选取原则:(一)血样:血浆(plasma)和血清(serum)是体内药物分析最常采用的样本,其中选用最多的是血浆。因血浆中的药浓可反映药物在体内(靶器管)的状况。而且血浆中药物浓度的数据报道较多,可供借鉴。血浆是全血(whole blood)在加肝素、枸橼酸、草酸盐等抗凝剂的全血经离心后分取,量约为全血的一半。血清则是在血液中纤维蛋白元等影响下,引起析出血块,离心取得。血块凝结时往往易造成药物吸附损失。全血也应加入抗凝剂混匀,以防凝血。对大多数药物来说血浆浓度与红细胞中的浓度成正比,所以测定全血也不能提供更多的数据,而全血的净化较血浆与血清麻烦,尤其是溶血后,血色素等可能会给测定带来影响。但是一些可与红血球结合或药物在血浆和血球的分配比率因不同病人而异的情况下,则宜采用全血。血样采取量会受到一定的限制,血样取样时间间隔问题也常随测定目的不同而异。目前大都是测定原型药物总量。当药物与血清蛋白结合率稳定时,血药总浓度可以有效表示游离药物的浓度。但对低蛋白症或尿毒症患者,药物结合率降低,则在通常安全有效的血药总浓度中,游离型药物浓度可显著增加。 (二)尿样(urine):尿样测定主要用于药物剂量回收研究、药物肾清除率和生物利用度等研究,以及测定代谢物类型等。体内药物清除主要是通过尿液排出,药物可以原型(母体药物)或代谢物及其缀合物形式排出。尿液药物浓度较高,收集量可以很大,但尿液浓度通常变化较大,所以宜测定一定时间内尿中药物的总量(如8、12、24小时内的累计量),需记录排出尿液体积及尿药浓度。尿药浓度改变不直接反映血药浓度,受试者肾功能将影响药物的排泄。尿中药物大多呈缀合状态,测定前要将缀合的药物游离。此外,采集尿液不可能在较短时间内多次取样,排尿时间较难掌握(尤其是婴儿),同时也具有不易采集完全的缺点。 (三)唾液(saliva):唾液中的药物浓度通常与血浆浓度相关。样品易得,取样无损害,尤易为儿童接收。有些可从药物唾液浓度推定血浆中游离药物浓度。但有些蛋白结合率较高的药物在唾液中的浓度比血浆浓度低得多,需高灵敏度的方法才能检测。唾液pH值6.9±0.5,每日分泌量1~1.5L,含有的主要电解质有Na+、K+、Cl-、HCO3-等,主要有机成分是粘[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]和淀粉酶。采样一般是在漱口后15分钟,收集口内自然流出或经舌在口内搅动后流出的混合唾液(吸管内吸附的少量唾液用稀释液洗出),用2000~3000rpm离心15分钟,小心吸取上清液,进一步分离、净化。也可采用物理(嚼石蜡片、小块聚四氟乙烯或玻璃大理石)或化学(酒石酸、维生素C)的方法刺激,在短时间内可得到大量唾液,但药浓也可能会受到影响。 (四)其它:乳汁、动物脏器组织匀浆等。 二、样品储存和稳定性考察:取样后最好立即进行分析,冷藏(4℃)、冰冻(-20℃)有时也不能完全保证样品不起变化。尿液是很好的细菌生长液,若需收集24小时或更长时间的样品或不能立即测定的,应置冰箱冷藏或加防腐剂(1%甲苯、过饱和氯仿)保存。分析样品贮存时应考虑:储存条件;样品在贮存中会对分析结果产生什么影响;评述样品稳定性时会发生什么问题;如何预防或校正不稳定样品的分析结果。 测定前样品的制备除少数体液经简单处理后直接测定外,通常在最后一步测定前要采取适当的样品制备,即进行分离、净化、浓集、必要时尚需对待测组分进行化学改性,为测定创造良好条件。一、样品的制备要考虑:药物的理化性质、待测物的浓度范围、药物测定的目的、选用的生物体液和组织的类型、样品制备与分析技术的关系。二、蛋白质的处理:是测定血浆、血清、全血及组织匀浆等样品中药物时的最先处理步骤。 (一)加入沉淀剂和变性试剂:硫酸铵是经典的蛋白质沉淀剂,它与蛋白质分子竞争系统中水分子,而使蛋白质析出。阴离子型沉淀剂(三氯醋酸、高氯酸、钨酸、焦磷酸)与带电荷的蛋白质在氏于等电点的pH时形成不溶性盐;反之,阳离子型沉淀剂(含锌盐、铜盐)与蛋白质分子中带阴电荷的羧基,在高于蛋白质等电点时,形成不溶性盐。有关机制不十分清楚。 (二)加入可与水混合的有机溶剂:乙醇过量存在时,能使与蛋白结合状态的药物释放可将混合物离心,取上清液(含药),但这不能解决样品的净化问题。蛋白沉淀法对于与蛋白结合力强的药物的回收率较差。也有采用酸消化法(Acid digestion)使药物自蛋白结合处释出,但常导致药物的分解。 (三)组织的酶消化法:蛋白水解酶(Proteolytic enzyme)中的枯草菌溶素(Subtilisin Carlsberg)不仅可使组织酶解,且可使药物析出。优点:1、因是在平稳条件下进行的,可避免某些药物在酸中水解及较高温度时降解;2、可显著改善对蛋白结合率强的药物的回收率;3、可用有机溶剂直接提取消化液而无乳化生成的危险;4、在用HPLC时,无需再进行过多的净化操作。缺点是不适用于一些在高pH时易水解的药物。三、提取:(一)溶液的pH调节:最佳pH选择主要与药物的pKa值有关。pH与pKa相当时,50%的药物以非电离形式存在。碱性药物最佳pH值要高于pKa值1~2个pH单位;反之,则低1~2个单位。可使90%药物以非电离开形式存在,易为溶剂提取。而对于碱性很强的药物往往采用“离子对”技术进行提取和定量。体内酸性物质较多,在碱性条件下不会被萃取出来,故在pH值偏高的情况下进行提取较好。 (二)提取溶剂的极性:选好第一个提取溶剂可减少以后的净化操作,在液-液提取中多采用极性小的溶剂。加入少量醇类可克服极性小溶剂提取能力弱和减小药物在容器表面的吸附损失的不足。也有利用不同极性的混合溶剂来提取药物和净化脂肪酸类。 (三)提取技术:由于体液样品量少且药物含量低,一次分析的样品数量较多。与常量和微量分析相比,提取时通常不采用反复提取的方法,多半进行一次(至多二次)提取,在改变pH后,从有机相回提至水相也只进行一次。一般并不考虑“提尽药物”,测定含量时则应精确加入提取溶剂,提取液也要定量分出。为避免进样时带来的误差,多采用提取前加入等量内标,以待测组分峰高(或峰面积)与内标峰高(或峰面积)之比对浓度作标准曲线。这样,即使在一系列操作过程中有微量损失,对比值影响也较小。混合时可在密塞情况下将试管平置于振荡器内振荡,振荡时间与强度视情况而定。可采用以“药物转入溶剂中量”与“混合时间”作图法,选取理想和符合实际的提取方式和时间。 (四)提取溶剂的蒸发:提取液常为数ml,往往不能直接供GC或HPLC测定,需采取浓集办法,常用真空蒸发(注意暴沸)或吹氮气流使溶剂挥散。四、固相分离:以固相分离方法进行样品预处理,从水相中分离出所需测定的组份,通常以柱分离方式进行操作,故有时这种方法又称为固相提取柱(Solid-phase extraction column)法。这类柱分两类:一种是阻留全部样品在柱上;一种则仅阻留药物及其相关物质。常用于填充柱的固相分离物质有氧化铝、活性碳、硅藻土、离子交换树脂及非离子型的树脂及凝胶等。在第一类柱中,常使用亲水性装填物,如硅藻土,可捕集全部样品。样品全部吸附在固相颗粒表面,形成一薄层,即使样品中含水也能如此。采用一种与水不相混溶的有机溶剂如氯乙烷倾入柱中,即可洗提药物。第二类柱则较有选择性,可装填疏水物或离子交换树脂,对药物及其相关物质进行滞留。常用的疏水物有活性碳、聚苯乙烯或C18化学键合硅胶。这种疏水柱可从样品中吸附亲脂性药物,然后用有机溶剂将药物洗提分离。离子交换柱适用于高极性、可电离的药物。五、利用分子大小进行分离—供血浆中游离药物的测定:采用超速离心、平衡透析、限外过滤(超滤)以及凝胶过滤方法等。以上方法也可用于药物蛋白结合率的测定。六、导致待测物损失的因素:(一)吸附:玻璃表面或橡胶塞会吸附药物,特别是脂肪胺类及含硫化合物。可采用硅烷化减少玻璃表面的吸附性,非极性提取溶剂中加入少量极性溶剂可减少器皿对药物的吸附。 血样中红血球和纤维蛋白元凝块的形成,常能引起待测物的共沉淀。所以宜将全血样品加入缓冲液后再行提







 我要推广仪器
我要推广仪器
 下载APP
下载APP