CFDA近两年药品、医疗器械法规文件及指导原则整理
p 2017年10月8日,中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,并发出通知。这是继2015年8月《国务院关于改革药品医疗器械审评审批制度的意见》之后,又一个深化药品医疗器械审评审批制度改革的纲领性文件,对我国医药产业创新发展具有里程碑意义。本文以《创新意见》为线索,对CFDA及CDE自2015年7月22日以来发布的重要文件进行梳理,为大家研究相关政策提供一份参考。 /p p span style=" color: rgb(0, 112, 192) " strong 1. 改革临床试验管理 /strong /span /p p 临床试验机构资格认定实行备案管理 /p p 支持临床试验机构和人员开展临床试验 /p p 完善伦理委员会机制 /p p 提高伦理审查效率 /p p 优化临床试验审批程序 /p p 接受境外临床试验数据 /p p 支持拓展性临床试验 /p p 严肃查处数据造假行为 /p p strong span style=" color: rgb(0, 112, 192) " 1.1 法规政策 /span /strong /p p 2016年3月23日,CFDA、卫计委委员会发布《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局 中华人民共和国国家卫生和计划生育委员会令第25号) /p p 2016年4月7日,CFDA发布关于贯彻实施《医疗器械临床试验质量管理规范》的通知(食药监办械管〔2016〕41号) /p p 2016年12月2日,总局办公厅公开征求《药物临床试验质量管理规范(修订稿)》的意见 /p p 2017年3月17日,总局公开征求《国家食品药品监督管理总局关于调整进口药品注册管理有关事项的决定(征求意见稿)》意见的通知 /p p 2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新改革临床试验管理的相关政策》(征求意见稿)意见的公告(2017年第53号) /p p 2017年8月4日,关于征求《医疗器械临床试验机构条件和备案管理办法(征求意见稿)》意见的函(食药监械管便函〔2017〕42号) /p p 2017年10月20日,CDE关于《接受境外临床试验数据的技术要求》征求意见通知 /p p strong span style=" color: rgb(0, 112, 192) " 1.2 指导原则 /span /strong /p p 2015年8月4日,CDE关于征求《儿科人群药物临床试验技术指导原则》意见的通知 /p p 2016年3月7日,总局关于发布儿科人群药物临床试验技术指导原则的通告(2016年第48号) /p p 2015年8月21日,CDE关于征求《药物临床试验的生物统计学指导原则》意见的通知 /p p 2016年6月3日,总局关于发布药物临床试验的生物统计学指导原则的通告(2016年第93号) /p p 2015年11月3日,国家食品药品监督管理总局关于发布中药新药临床研究一般原则等4个技术指导原则的通告(2015年第83号) /p p 2015年12月24日,CDE关于《药物临床试验的一般考虑》指导原则征求意见的通知 /p p 2017年1月20日,总局关于发布药物临床试验的一般考虑指导原则的通告(2017年第11号) /p p 2016年1月29日,关于《临床试验数据管理工作技术指南》、《临床试验的电子数据采集(EDC)技术指导原则》和《药物临床试验数据管理和统计分析的计划和报告指导原则》征求意见的通知 /p p 2016年7月29日,总局关于发布临床试验的电子数据采集技术指导原则的通告(2016年第114号) /p p 2016年7月29日,总局关于发布药物临床试验数据管理与统计分析的计划和报告指导原则的通告(2016年第113号) /p p 2016年7月29日,总局关于发布临床试验数据管理工作技术指南的通告(2016年第112号) /p p 2016年9月30日,CDE关于征求《新药I期临床试验申请技术指南(草案)》意见的通知 /p p 2016年10月11日,总局关于发布中药新药治疗流行性感冒临床研究技术指导原则的通告(2016年第136号) /p p 2016年10月29日,CDE关于征求《成人用药数据外推在儿科人群药物临床试验及相关信息使用的技术指导原则》意见的通知 /p p 2017年5月18日,总局关于发布成人用药数据外推至儿科人群的技术指导原则的通告(2017年第79号) /p p 2017年7月3日,关于《创新药(化学药)Ⅲ期临床试验药学研究信息指南(草案)》征求意见的通知 /p p strong span style=" color: rgb(0, 112, 192) " 2. 加快上市审评审批 /span /strong /p p 加快临床急需药品医疗器械审评审批 /p p 支持罕见病治疗药品医疗器械研发 /p p 严格药品注射剂审评审批 /p p 实行药品与药用原辅料和包装材料关联审批 /p p 支持中药传承和创新 /p p 建立专利强制许可药品优先审评审批制度 /p p strong span style=" color: rgb(0, 112, 192) " 2.1 法规政策 /span /strong /p p 2015年11月13日,国家食品药品监督管理总局关于征求《关于解决药品注册申请积压实行优先审评审批的意见(征求意见稿)》意见的公告(2015年第227号) /p p 2015年12月24日,总局关于征求《中药配方颗粒管理办法(征求意见稿)》意见的公告(2015年第283号) /p p 2015年12月31日,国家食品药品监督管理总局关于落实中药提取和提取物监督管理有关规定的公告(2015年第286号) /p p 2016年2月26日,CFDA发布关于解决药品注册申请积压实行优先审评审批的意见(食药监药化管〔2016〕19号) /p p 2016年3月18日,总局关于取消中药材生产质量管理规范认证有关事宜的公告(2016年第72号) /p p 2016年1月12日,总局关于征求药包材和药用辅料关联审评审批申报资料要求(征求意见稿)意见的公告(2016年第3号) /p p 2016年11月28日,总局关于发布药包材药用辅料申报资料要求(试行)的通告(2016年第155号) /p p 2016年5月12日,总局办公厅公开征求关于药包材药用辅料与药品关联审评审批有关事项的公告(征求意见稿)意见 /p p 2016年8月10日,总局关于药包材药用辅料与药品关联审评审批有关事项的公告(2016年第134号) /p p 2016年6月21日,关于征求医疗器械优先审批程序意见的函(食药监械管便函〔2016〕40号) /p p 2016年10月26日,总局关于发布医疗器械优先审批程序的公告(2016年第168号) /p p 2016年7月21日,CDE关于征求《“首仿”品种实行优先审评评定的基本原则》的意见 /p p 2017年7月21日,总局办公厅公开征求《关于对医疗机构应用传统工艺配制中药制剂实施备案管理的公告(征求意见稿)》意见 /p p 2017年10月9日,总局办公厅公开征求《中药经典名方复方制剂简化注册审批管理规定(征求意见稿)》及申报资料要求(征求意见稿)意见 /p p strong span style=" color: rgb(0, 112, 192) " 2.2 指导原则 /span /strong /p p 2017年1月11日,总局办公厅公开征求中成药通用名称命名技术指导原则(征求意见稿)的意见 /p p 2017年2月16日,总局关于发布医疗器械优先审批申报资料编写指南(试行)的通告(2017年第28号) /p p 2017年3月3日,CDE关于征求《儿科用药非临床安全性研究技术指导原则》意见的通知 /p p 2017年3月28日,CDE关于再次征求《新药I期临床试验申请技术指南》意见的通知 /p p 2017年4月13日,CDE关于6个中药新药临床研究技术指导原则上网征求意见的通知 /p p 2017年10月11日,总局办公厅公开征求《中药资源评估技术指导原则》意见 /p p 2017年10月11日,总局办公厅公开征求《中成药规格表述技术指导原则(征求意见稿)》意见 /p p strong span style=" color: rgb(0, 112, 192) " 3. 促进药品创新和仿制药发展 /span /strong /p p 建立上市药品目录集 /p p 探索药品专利链接制度 /p p 开展药品专利期限补偿制度试点 /p p 完善和落实药品试验数据保护制度 /p p 促进药品仿制生产 /p p 发挥企业的创新主体作用 /p p 支持新药临床应用 /p p strong span style=" color: rgb(0, 112, 192) " 3.1 创新药 /span /strong /p p strong span style=" color: rgb(0, 112, 192) " 3.1.1 法规政策 /span /strong /p p 2015年11月6日,总局关于征求药品上市许可持有人制度试点方案和化学药品注册分类改革工作方案两个征求意见稿意见的公告(2015年 第220号) /p p 2016年3月9日,总局关于发布化学药品注册分类改革工作方案的公告(2016年第51号) /p p 2016年5月4日,总局关于发布化学药品新注册分类申报资料要求(试行)的通告(2016年 第80号) /p p 2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新加快新药医疗器械上市审评审批的相关政策》(征求意见稿)意见的公告(2017年第52号) /p p 2017年5月12日,总局关于征求《关于鼓励药品医疗器械创新保护创新者权益的相关政策(征求意见稿)》意见的公告(2017年第55号) /p p 3.1.2 指导原则 /p p 2017年5月30日,药品审评中心关于征求《药品电子通用技术文档结构(征求意见稿)》意见的通知 /p p 2017年10月17日,药品审评中心关于再次征求《药品电子通用技术文档结构》意见的通知 /p p strong span style=" color: rgb(0, 112, 192) " 3.2 仿制药 /span /strong /p p strong span style=" color: rgb(0, 112, 192) " 3.2.1 法规政策 /span /strong /p p 2015年11月6日,国家食品药品监督管理总局关于征求化学仿制药生物等效性试验备案管理规定(征求意见稿)意见的公告(2015年第221号) /p p 2015年11月18日,国家食品药品监督管理总局关于征求《关于开展仿制药质量和疗效一致性评价的意见(征求意见稿)》意见的公告(2015年 第231号) /p p 2016年3月5日,国务院办公厅印发关于开展仿制药质量和疗效一致性评价的意见(国办发〔2016〕8号) /p p 2016年3月10日,总局办公厅公开征求研制过程中所需研究用对照药品一次性进口有关事宜的意见 /p p 2016年7月1日,总局关于研制过程中所需研究用对照药品一次性进口有关事宜的公告(2016年第120号) /p p 2016年3月28日,总局办公厅公开征求仿制药质量和疗效一致性评价工作程序及化学药品仿制药口服固体制剂一致性评价申报资料要求意见 /p p 2016年8月17日,总局关于发布化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)的通告(2016年第120号) /p p 2016年4月1日,总局办公厅公开征求关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》的有关事项的意见 /p p 2016年5月26日,总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的公告(2016年第106号) /p p 2016年5月26日,总局关于发布仿制药质量和疗效一致性评价工作程序的公告(2016年第105号) /p p 2016年4月12日,食品药品监管总局办公厅公开征求仿制药质量和疗效一致性评价参比制剂备案与推荐程序的意见 /p p 2016年5月19日,总局关于发布仿制药质量和疗效一致性评价参比制剂备案与推荐程序的公告(2016年第99号) /p p 2017年6月9日,总局办公厅公开征求《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》意见 /p p 2017年8月25日,总局关于仿制药质量和疗效一致性评价工作有关事项的公告(2017年第100号) /p p 2017年9月4日,CDE关于公开征求《中国上市药品目录集》框架意见的通知 /p p 2017年9月22日,仿制药质量与疗效一致性评价办公室关于发布《仿制药质量和疗效一致性评价申报资料立卷审查技术标准(暂行)》的通知 /p p 2017年10月13日,国家食品药品监督管理总局、国家卫生和计划生育委员会关于药物临床试验机构开展人体生物等效性试验的公告(2017年第119号) /p p strong span style=" color: rgb(0, 112, 192) " 3.2.1 指导原则 /span /strong /p p 2015年10月30日,1.普通口服固体制剂参比制剂选择和确定指导原则(征求意见稿) 2.普通口服固体制剂溶出曲线测定与比较指导原则(征求意见稿) 3.仿制药质量一致性评价人体生物等效性研究技术指导原则(征求意见稿) /p p 2016年3月18日,总局发布了《普通口服固体制剂参比制剂选择和确定指导原则》《普通口服固体制剂溶出曲线测定与比较指导原则》《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》 /p p 2015年11月27日,食品药品监管总局办公厅关于征求化学仿制药CTD格式申报资料撰写要求意见的通知(食药监办药化管函〔2015〕737号) /p p 2015年11月27日,CDE《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》上网征求意见的通知 /p p 2016年4月8日,总局办公厅公开征求人体生物等效性试验豁免指导原则的意见 /p p 2016年5月19日,总局关于发布人体生物等效性试验豁免指导原则的通告(2016年第87号) /p p 2016年9月13日,总局办公厅公开征求仿制药质量和疗效一致性评价改规格药品评价一般考虑的意见 /p p 2016年9月14日,总局办公厅公开征求仿制药质量和疗效一致性评价临床有效性试验一般考虑的意见 /p p 2017年2月7日,总局关于发布仿制药质量和疗效一致性评价临床有效性试验一般考虑的通告(2017年第18号) /p p 2016年11月7日,总局办公厅公开征求仿制药质量和疗效一致性评价工作中改剂型药品(普通口服固体制剂)评价一般考虑的意见 /p p 2016年11月7日,总局办公厅公开征求仿制药质量和疗效一致性评价工作中改盐基药品评价一般考虑的意见 /p p 2016年11月29日,总局办公厅公开征求进一步规范仿制药质量和疗效一致性评价参比制剂选择等相关事宜的指导意见的意见 /p p 2016年11月29日,总局办公厅公开征求仿制药质量和疗效一致性评价品种分类的指导意见的意见 /p p 2017年4月5日,总局关于发布仿制药质量和疗效一致性评价品种分类指导意见的通告(2017年第49号) /p p 2016年12月21日,总局办公厅公开征求仿制药质量和疗效一致性评价研究现场核查等指导原则的意见 /p p 2017年5月18日,总局关于发布仿制药质量和疗效一致性评价研制现场核查指导原则等4个指导原则的通告(2017年第77号) /p p 2017年4月28日,总局办公厅公开征求化学仿制药口服固体制剂一致性评价复核检验技术指南(征求意见稿)的意见 /p p 2017年5月18日,总局办公厅公开征求胃肠道局部作用药物、电解质平衡用药仿制药质量和疗效一致性评价及特殊药品生物等效性试验申请有关事宜意见(征求意见稿)的意见 /p p 2017年5月25日,总局办公厅公开征求胃肠道局部作用药物、电解质平衡用药仿制药质量和疗效一致性评价及特殊药品生物等效性试验申请有关事宜意见(征求意见稿)的意见 /p p 2017年5月30日,药品审评中心关于征求《化学仿制药电子通用技术文档申报指导原则(征求意见稿)》意见的通知 /p p 2017年10月17日,药品审评中心关于再次征求《化学仿制药电子通用技术文档申报指导原则》意见的通知 /p p 2017年6月9日,总局办公厅公开征求《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)(征求意见稿)》《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)(征求意见稿)》及相关单据意见 /p p 2017年9月5日,总局关于发布《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)》《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)》的通告(2017年第148号) /p p strong span style=" color: rgb(0, 112, 192) " 4. 加强药品医疗器械全生命周期管理 /span /strong /p p 推动上市许可持有人制度全面实施 /p p 落实上市许可持有人法律责任 /p p 建立上市许可持有人直接报告不良反应和不良事件制度 /p p 开展药品注射剂再评价 /p p 完善医疗器械再评价制度 /p p 规范药品学术推广行为 /p p strong span style=" color: rgb(0, 112, 192) " 4.1 法规政策 /span /strong /p p 2015年11月6日,总局关于征求药品上市许可持有人制度试点方案和化学药品注册分类改革工作方案两个征求意见稿意见的公告(2015年 第220号) /p p 2015年11月12日,食品药品监管总局办公厅、国家卫生计生委办公厅关于公开征求医疗器械不良事件监测和再评价管理办法(征求意见稿)意见的函(食药监办械监函〔2015〕723号) /p p 2016年2月20日,总局公开征求对药品经营质量管理规范修订的意见 /p p 2016年5月6日,关于征求《医疗器械冷链(运输、贮存)管理指南》意见的函(食药监械监便函〔2016〕61号) /p p 2016年9月22日,总局关于发布医疗器械冷链(运输、贮存)管理指南的公告(2016年第154号) /p p 2016年6月6日,国务院办公厅发布关于印发药品上市许可持有人制度试点方案的通知(国办发〔2016〕41号) /p p 2016年7月7日,CFDA发布关于做好药品上市许可持有人制度试点有关工作的通知(食药监药化管〔2016〕86号) /p p 2016年9月31日,关于征求《医疗器械生产企业质量控制与成品放行指南(征求意见稿)》意见的函(食药监械监便函〔2016〕127号) /p p 2016年10月31日,总局关于公开征求医疗器械不良事件监测和再评价管理办法(征求意见稿)意见的通知 /p p 2017年2月8日,CFDA发布《医疗器械召回管理办法》(国家食品药品监督管理总局令第29号) /p p 2017年5月11日,总局关于征求《关于鼓励药品医疗器械创新实施药品医疗器械全生命周期管理的相关政策》(征求意见稿)意见的公告(2017年第54号) /p p 2017年6月21日,总局关于公开征求《网络医疗器械经营监督管理办法(征求意见稿)》意见的通知 /p p 2017年10月17日,总局办公厅公开征求《药品生产场地变更简化注册审批管理规定(征求意见稿)》及《药品生产场地变更研究技术指导原则(征求意见稿)》意见 /p p 2017年10月27日, 总局办公厅公开征求《中药材生产质量管理规范(修订稿)》意见 /p p strong span style=" color: rgb(0, 112, 192) " 4.2 指导原则 /span /strong /p p 2017年1月10日,CDE关于《已上市化学药品生产工艺变更研究技术指导原则》征求意见的通知 /p p 2017年8月29日,总局关于发布已上市化学药品生产工艺变更研究技术指导原则的通告(2017年第140号) /p p 2017年3月6日,CDE关于《已上市中药生产工艺变更研究技术指导原则》征求意见的通知 /p p 2017年8月24日,总局关于发布已上市中药生产工艺变更研究技术指导原则的通告(2017年第141号) /p p strong span style=" color: rgb(0, 112, 192) " 5. 提升技术支撑能力 /span /strong /p p 完善技术审评制度 /p p 落实相关工作人员保密责任 /p p 加强审评检查能力建设 /p p 落实全过程检查责任 /p p 加强国际合作 /p p strong span style=" color: rgb(0, 112, 192) " 5.1 配套支持 /span /strong /p p 2015年12月10日,国家食品药品监督管理总局关于征求《生物制品批签发管理办法》(修订稿)意见的公告(2015年第263号) /p p 2016年12月13日,总局公开征求生物制品批签发管理办法修订草案意见的通知 /p p 2016年5月31日,总局关于药物非临床研究质量管理规范认证和药物临床试验机构资格认定施行电子申请受理的公告(2016年第110号) /p p 2016年6月6日,总局关于发布药物研发与技术审评沟通交流管理办法(试行)的通告(2016年第94号) /p p 2016年7月25日,总局办公厅公开征求《药品注册管理办法(修订稿)》意见 /p p 2017年10月23日,总局办公厅公开征求《药品注册管理办法(修订稿)》意见 /p p 2016年8月19日,总局办公厅公开征求《药物非临床研究质量管理规范(修订稿)》意见 /p p 2017年8月2日,CFDA发布《药物非临床研究质量管理规范》(国家食品药品监督管理总局令第34号) /p p 2016年9月20日,总局办公厅公开征求《进口药材管理办法(修订稿)》意见 /p p 2016年9月21日,关于征求《体外诊断试剂注册管理办法》修正案意见的函(食药监械管便函〔2016〕62号) /p p 2016年9月30日,总局办公厅关于征求医疗器械分类目录(修订稿)意见的函 /p p 2017年9月4日,总局关于发布医疗器械分类目录的公告(2017年第104号) /p p 2017年9月4日,总局关于实施《医疗器械分类目录》有关事项的通告(2017年第143号) /p p 2016年10月10日,食品药品审核查验中心公开征求《药品数据管理规范》的意见 /p p 2017年8月25日,总局办公厅公开征求《药品数据管理规范(征求意见稿)》意见 /p p 2016年11月22日,总局办公厅公开征求药品标准管理办法(征求意见稿)意见 /p p 2016年10月31日,总局关于公开征求《医疗器械标准管理办法》(征求意见稿)意见的通知 /p p 2016年12月6日,CDE关于《药品审评项目管理办法》征求意见的通知 /p p 2016年12月29日,总局关于调整部分行政审批事项审批程序决定公开征求意见的通知 /p p 2017年4月5日,CFDA发布《国家食品药品监督管理总局关于调整部分药品行政审批事项审批程序的决定》(国家食品药品监督管理总局令第31号) /p p 2017年4月6日,CFDA发布《国家食品药品监督管理总局关于调整部分医疗器械行政审批事项审批程序的决定》(国家食品药品监督管理总局令第32号) /p p 2016年12月29日,总局关于《国家食品药品监督管理总局药品医疗器械审评审批保密管理办法﹙征求意见稿﹚》公开征求意见的通知 /p p 2017年2月23日,总局办公厅公开征求《关于药品再注册有关事项的公告(征求意见稿)》意见 /p p 2017年3月9日,总局关于发布药品注册审评专家咨询委员会管理办法(试行)的公告(2017年第27号) /p p 2017年8月31日,总局办公厅公开征求《药品境外检查规定(征求意见稿)》意见 /p p 2017年9月13日,总局办公厅公开征求《关于调整药品注册受理工作的公告(征求意见稿)》意见 /p p 2017年10月23日,总局办公厅公开征求《〈中华人民共和国药品管理法〉修正案(草案征求意见稿)》意见 /p p br/ /p











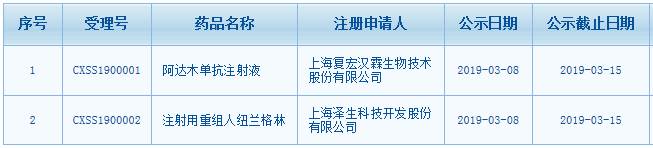











 我要推广仪器
我要推广仪器
 下载APP
下载APP