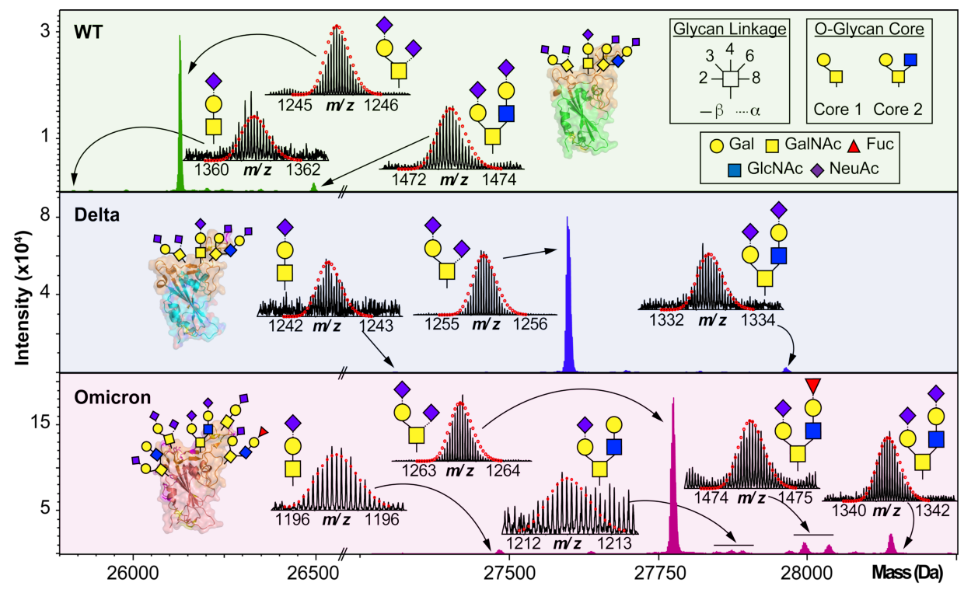
布鲁克:基于捕集型离子淌度质谱的4D-蛋白质组学
p style=" text-align: left background: rgb(255, 255, 255) margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 蛋白质组学是对复杂生物样品中蛋白质结构和功能的大规模系统性研究,生物样本的高复杂性和不均一性为蛋白质组学研究带来了极大挑战。近年来,离子淌度与高分辨质谱的联合使用,使得蛋白质组学进入了4D新时代。4D-蛋白质组学是指在3D分离即保留时间(retention time)、质荷比(m/z)、离子强度(intensity)这三个维度的基础之上,增加了第四个维度--离子淌度(mobility),根据离子的形态、大小进行分离(图1)。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " img style=" max-width: 100% max-height: 100% width: 600px height: 305px " src=" https://img1.17img.cn/17img/images/202007/uepic/7b2a33cb-0815-40c6-b214-afc66996233a.jpg" title=" 图片1.png" alt=" 图片1.png" width=" 600" height=" 305" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-align: center text-indent: 0em " 图1. 新一代4D-蛋白质组学示意图 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 布鲁克推出的基于timsTOF Pro的4D-蛋白组学平台,采用了创新的TIMS(Trapped Ion Mobility Spectrometry,捕集离子淌度)技术和PASEF(Parallel Accumulation Serial Fragmentation,平行累积连续碎裂)采集模式(图2)。捕集型离子淌度TIMS是指离子在气流的推动下向前运动,将离子按大小和形状分开,在离子运动的反方向施加电场,阻滞离子的运动,将离子trap在特定的位置,然后逐渐降低电场,将离子由大到小逐个释放。timsTOF Pro使用了双TIMS结构,具有独特的PASEF扫描模式,离子在第一个TIMS部分中进行累积并聚焦,然后传输到第二个TIMS中,进行淌度分离和释放;同时第一段TIMS会重新进行新一批离子的累积;并且,四级杆对母离子的选择、碰撞池对离子的碎裂与TIMS中离子的释放同步进行,实现快速、高效的二级采集。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 279px " src=" https://img1.17img.cn/17img/images/202007/uepic/60853c3f-0b18-48df-8afc-114acc2bc4ab.jpg" title=" 图片2.png" alt=" 图片2.png" width=" 600" height=" 279" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图2. timsTOF Pro的结构示意图 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 基于timsTOF Pro的4D-蛋白组学平台,具有鉴定深度、定量准确性、检测速度、仪器稳定性等性能的全面提升: /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 具有色谱保留时间、离子淌度、质荷比、谱峰强度4维信息,显著提高对复杂样本的分离能力和谱图质量,促进了共流出组分的同分异构体区分; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 创新双TIMS设计,使离子的累积和淌度分离同步进行,带来近100%的Duty Cycle; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 离子碰撞截面积(CCS)值的准确、高重现性测量; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 灵敏度的革命性提升,适合微量样本的组学分析; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 超过100 Hz二级扫描速度; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 超级稳定的整体设计,能够保证长时间连续样本测试的稳定性,耐用性,易维护。 span style=" text-indent: 2em " & nbsp /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 基于timsTOF Pro的4D-蛋白组学平台不仅适合于蛋白质组的深度鉴定、定量分析,还适合于翻译后修饰的精准研究,临床大队列样本的快速检测,微量样本甚至单细胞蛋白质组研究、蛋白质复合体交联分析等。该平台发布两年多以来,已经得到越来越多的蛋白组学研究团队认可并开始使用此革命性技术,一方面,是因为过去两年多已经有大量的数据证明,timsTOF Pro的采集速度和灵敏度的同时提升大大突破了蛋白组学研究现有瓶颈,这提高了基础蛋白组学研究的水平;另一方面,timsTOF Pro灵敏度和扫描速度上的独特优势,意味着所以可以用更低的进样量和更短的色谱梯度鉴定到更多的蛋白,同时离子淌度的引入,更是大幅提高了数据的完整性和谱图的归属性,这些特点对基础蛋白组学研究向临床蛋白组学应用的转化至关重要。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " span style=" color: rgb(79, 129, 189) " strong 4D-蛋白质组学技术提高蛋白和多肽覆盖深度 /strong /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " strong 基于质谱技术的蛋白质组学研究方法在生命科学研究的各个领域都取得了傲人的成果,但由于蛋白质组学样本的自身复杂性(蛋白丰度的动态范围& gt 106)和目前质谱仪采集速度和灵敏度的局限性,低丰度蛋白鉴定异常困难,这让深度覆盖蛋白组学面临着巨大的挑战,提高蛋白质组学覆盖深度一直是科研工作者努力的方向之一。 /strong /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 4D组学质谱平台timsTOF Pro的出现,让蛋白组学技术产生了革命性的变化,timsTOF Pro采用双TIMS结构,并采用独特的PASEF扫描模式,可以提供超过100 Hz的MSMS扫描速度,能使二级采集速度和灵敏度同时提高,这个特性可以完美应对传统质谱在采集速度和灵敏度方面的挑战。timsTOF Pro出色的灵敏度,只需要传统分析十分之一的进样量,就可以得到更好的鉴定深度(图3),这让timsTOF Pro更加适合生物标志物研究、药物发现、临床蛋白组学研究和单细胞蛋白组学等这些样本量通常会比较少的应用。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 258px " src=" https://img1.17img.cn/17img/images/202007/uepic/c49572a1-0f8c-4b8e-ad7e-510ac1369573.jpg" title=" 图片3.png" alt=" 图片3.png" width=" 600" height=" 258" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图3. timsTOF Pro的蛋白水平和多肽水平深度覆盖 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " span style=" color: rgb(79, 129, 189) " strong 4D-蛋白质组学技术带来更精确的翻译后修饰组学研究 /strong /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 蛋白质翻译后修饰(如磷酸化、糖基化、甲基化、乙酰化和泛素化等)通过动态调控蛋白的结构和功能,参与信号传导、基因表达、物质代谢等多种生命活动,成为了科研工作的关注焦点。近年来,随着样品制备手段和质谱技术的快速发展,翻译后修饰的研究方法不断涌现。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 传统的质谱分析技术在蛋白质翻译后修饰研究中常面临着巨大的挑战: /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 蛋白质的翻译后修饰在样本中含量低且动态范围广; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 应用于蛋白质翻译后修饰的研究策略主要还是基于鸟枪法的蛋白组学,酶切极大提高了样本复杂度; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 由修饰位点不同带来的同分异构多肽会在色谱上存在严重的共洗脱问题,而这些同分异构肽段在传统蛋白组学质谱平台上不能得到有效分离。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 由于翻译后修饰分布广泛且含量较低,往往需要进行修饰肽段的富集,所得到的样本量较少,需要灵敏度更高的仪器进行检测;此外,翻译后修饰位点、修饰类型的确认,对于蛋白功能的解析至关重要。布鲁克推出的4D-蛋白组学平台timsTOF Pro,提高了峰容量和修饰位点异构鉴定的可信度,具有大于100Hz的扫描速度和优越的灵敏度,显著提高了翻译后修饰的鉴定深度和位点鉴定的准确性(图4)。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 265px " src=" https://img1.17img.cn/17img/images/202007/uepic/5bfed120-716a-48f7-b0e9-9801dd628a74.jpg" title=" 图片4.png" alt=" 图片4.png" width=" 600" height=" 265" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em " 图4. 磷酸化组学分析。A.& nbsp 50-100 ug起始蛋白量进行IMAC富集,采用不同色谱梯度,单针分析磷酸化多肽鉴定数目。B. 离子淌度可以准确区分修饰位点异构,提高修饰鉴定和定量的可靠性。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " span style=" color: rgb(79, 129, 189) " strong 4D-蛋白质组学加快组学研究向临床应用转化 /strong /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 蛋白质组学不仅是研究生命活动、疾病机理的最有效方法之一,而且在对癌症、老年痴呆等人类重大疾病的分子诊断和临床治疗方面也有十分广阔的前景。随着样本制备、色谱分离和质谱技术的进步,临床蛋白组学渐渐开始走向大队列研究,矩形研究策略则是趋势。高通量蛋白组学则成为了生物医学基础研究和应用开发的重要前沿和突破口,而如何实现对大队列样本稳定可靠地分析也逐渐成为了科研热点和难点。总的来说,实现高通量临床蛋白组学面临如下挑战: /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 蛋白质组学样本自身的复杂性和不均一性(蛋白丰度的动态范围& gt 106),使得低丰度蛋白鉴定和定量异常困难; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 目前质谱仪采集速度和灵敏度的局限性,使得短梯度下难以实现蛋白深度覆盖; /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp 大队列样本分析对高通量方法和仪器稳定性提出了很高的要求。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 基于timsTOF Pro的4D-蛋白组学平台具有更快的扫描速度、更高的灵敏度和更好的离子选择性,显现出了向临床转化的广阔前景。布鲁克应用专家以及timsTOF Pro的用户做了大量工作,开发了多个成熟的高通量样本检测的应用方案,以探索蛋白组学技术用于临床研究的可能。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 布鲁克nanoElute LC为timsTOF Pro质谱标配的纳升流液相,采用短梯度色谱方法(图5A)28.8min一个循环,单针进样200ng的HeLa平均可以鉴定4180种蛋白质(图5B),26000条多肽(图5C),30针重复的相关系数R& gt 0.97(图5D)。这些结果表明,此方法与timsTOF Pro的高扫描速度和灵敏度结合,能同时兼顾分析通量和蛋白覆盖深度。我们将此方法用于多组份样本分析,小于12小时即可完成24个组份分析(图5E),HeLa样本24个组份可以鉴定大于9000种蛋白质,小鼠小脑样本24个组份可以鉴定大于10000种蛋白质。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 359px " src=" https://img1.17img.cn/17img/images/202007/uepic/999652fb-442b-4b16-bebd-29d5867ff800.jpg" title=" 图片5.png" alt=" 图片5.png" width=" 600" height=" 359" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图5. 基于nanoElute短梯度高通量方案 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " span style=" text-indent: 2em " 布鲁克与Evosep公司合作,联合推出用于临床蛋白质组学研究的整体解决方案,timsTOF Pro出色的扫描速度和灵敏度与Evosep One LC强大的分离能力和稳定性完美适配。英国牛津大学纳菲尔德医学系Roman Fischer教授,采用这套系统进行临床大队列的的血液蛋白组研究,用于快速发现疾病标志物。将收集的192例血浆样本去除12个高丰度蛋白,在timsTOF Pro与Evosep液相平台上使用11.5分钟梯度(100例样品/天)分析,样本测试中插入20针QC样本进行质控,总计212个样本仅需51小时测试时间(图6A),这项工作采用传统的质谱方案可能需要接近10天。实验结果显示,采用4D Match Between Runs,192个样本中,可以对500个蛋白进行定量分析,并且CV值小于10%,而QC样本的CV值小于5%(图6B、6C)。 /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " & nbsp /p p style=" text-align: center text-indent: 0em " img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202007/uepic/7c042971-4e8a-4eca-ae3c-d7e819e5b0f9.jpg" title=" 图片6.png" alt=" 图片6.png" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图6. & nbsp Roman Fischer教授采用的临床血液蛋白组研究案例 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " & nbsp /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 创新的4D-DIA非标记定量技术:dia-PASEF@ /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 布鲁克与苏黎世联邦理工学院、德国马普研究所和多伦多大学合作,将PASEF与DIA(Data-Independent Acquisition,数据非依赖采集)的优势相结合,产生了一种新的采集模式称为dia-PASEF@(图7)。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " & nbsp /p p style=" text-align: center text-indent: 0em " img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202007/uepic/5d5aae78-f80d-488e-a9e9-da3eab36fa54.jpg" title=" 图片7.png" alt=" 图片7.png" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图7. 全新dia-PASEF@采集策略 span style=" text-indent: 2em " & nbsp /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 在dia-PASEF@扫描模式中,母离子在进入四级杆之前已经通过淌度进行了累积和分离,根据碰撞截面积(CCS)大小依次洗脱(与m/z有一定相关性),这样四级杆就可以根据洗脱离子的m/z进行离子选择,并且每批PASEF都会有多个窗口进行扫描,从而提高离子的利用率,避免了传统DIA方法中离子利用率低的问题。此外,使用离子淌度和质量数双重隔离窗口来触发MS/MS,提高了母离子选择和匹配的准确性,并降低了二级混合谱图的复杂性。并且在一级热图中,可以选择多电荷区域作为dia-PASEF@的母离子窗口,有效屏蔽单电荷杂质的干扰。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 306px " src=" https://img1.17img.cn/17img/images/202007/uepic/df424259-85f1-478c-8cd3-dc81f106d619.jpg" title=" 图片8.png" alt=" 图片8.png" width=" 600" height=" 306" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图8. dia-PASEF@进行高通量、微量样本的蛋白质组定量分析 span style=" text-indent: 0em " & nbsp /span /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " dia-PASEF@具有更深的蛋白质组覆更高的分析通量和更优异的灵敏度,适合于高通量、微量样本的蛋白质组定量分析。采用95min梯度,单针进样200ng的HeLa,利用dia-PASEF@可以鉴定超过7,600种蛋白质、66,000种肽段。采用不同长度的梯度,30min可以鉴定6395个Hela细胞蛋白、4032个Yeast蛋白,达到快速、深度覆盖。即使在微量样本时,如单针进样10ng的Hela,仍能鉴定3000种蛋白质。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 布鲁克在ASMS 2020发布了高通量dia-PASEF@方案(图9A),即将Evosep One LC与timsTOF Pro再次联合,最大程度发挥Evosep One LC快速分离和timsTOF Pro扫描速度和稳定性的优势。该方案目前有4种方法(图9B),分别采用4.8min、7.2min、14.4min和24min色谱方法,对应的每天可以分析300、200、100和60蛋白质组学样本,把蛋白组学分析通量提升到一个全新的高度。分析结果(图9C,9D)显示出此方案在保证分析通量的同时,蛋白覆盖深度也有很好的保证,4.8min的分析单针可以鉴定2158蛋白,24min可以得到与传统蛋白组学长梯度分析相当的结果。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 284px " src=" https://img1.17img.cn/17img/images/202007/uepic/fb3f5696-51bb-453a-8e70-cf3ba53b5151.jpg" title=" 图片9.png" alt=" 图片9.png" width=" 600" height=" 284" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图9. 高通量dia-PASEF@方案 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 英国牛津大学Roman Fischer教授采用高通量dia-PASEF@方案,进行血液蛋白质组学大队列研究,43天完成4300针连续进样,总共采集2.5亿张二级谱图,整个采集过程只需一次离子传输管清洗(图10)。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 324px " src=" https://img1.17img.cn/17img/images/202007/uepic/4ca80514-f97a-4b36-b172-fa4ce1cf4a03.jpg" title=" 图片10.png" alt=" 图片10.png" width=" 600" height=" 324" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图10. Roman Fischer教授采用dia-PASEF@技术进行大队列研究 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 创新的4D-PRM靶向定量技术:prm-PASEF@ /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 在布鲁克的革命性timsTOF Pro平台上,通过将PASEF与平行反应监测(PRM)相结合,使其非标记靶向蛋白质组学性能得到了进一步增强。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " prm-PASEF@技术在保持高灵敏度的同时,使靶向离子数目最大化,100ms的PASEF扫描时间可以靶向10个以上的肽段,有效离子利用率提高10倍以上,实现了谱峰上采集点数目最大化,显著提高数据的完整性(图11A)。prm-PASEF@技术根据色谱流出时间、淌度、质荷比以及碎片离子的分辨能力进行离子筛选,具有更好的选择性,定量更加准确(图11B)。此外,来自卢森堡健康研究所Antoin Lesur教授采用prm-PASEF@技术在细胞样本里30min梯度检测213种靶向肽段,在5amol到50fmol范围都可以准确定量,具有优秀的灵敏度(图11C)。 /p p style=" text-align: center text-indent: 0em " img style=" max-width: 100% max-height: 100% width: 600px height: 428px " src=" https://img1.17img.cn/17img/images/202007/uepic/4aa99cb0-4e1d-46da-87c1-f588c4faa99a.jpg" title=" 图片11.png" alt=" 图片11.png" width=" 600" height=" 428" border=" 0" vspace=" 0" / /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 0em text-align: center " 图11. prm-PASEF@进行靶向蛋白质组定量分析 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 综上,捕集型离子淌度技术引领蛋白质组学进入4D新时代,基于timsTOF Pro的4D-蛋白质组学平台在蛋白质组分析方面展现出极佳的灵敏度、采集速度和覆盖深度,有利于实现更精确的翻译后修饰分析、高通量的临床大队列样本分析。新开发的dia-PASEF@和prm-PASEF@离子利用率高,具有更高的鉴定深度和定量准确性。随着布鲁克和合作团队对蛋白组学方案的不断开发,4D-蛋白质组学必将越来越完善,展现出更加广泛的应用前景。 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " & nbsp & nbsp /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " 参考文献 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Antoine Lesur, et al.,& nbsp New prm-PASEF for highly multiplexed targeted acquisition in clinical samples. ASMS 2020, Poster TP470. /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Christopher M. Adams, et al.,& nbsp Identification and Quantitation of Phosphopeptide PositionalIsomers using Trapped Ion Mobility Spectrometry and PASEF. ASMS 2019, WP 662 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Florian Meier,& nbsp et al.,& nbsp Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Molecular & amp Cellular Proteomics, 2018,17(12),2534-2545. /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Florian Meier,& nbsp et al.,& nbsp Parallel Accumulation–Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14,12, 5378-5387 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Matthew Willetts,et al.,& nbsp High Sensitivity PTM Characterization in Complex Cell Lysates Using Trapped Ion Mobility. ASMS 2019, Poster TP630 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Shourjo Ghose,et al.,& nbsp Analysis of Histones from HEK293T Cells using a QTOF with Trapped Ion Mobility and PASEF Workflows. ASMS 2019, Poster TP642 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Stephanie Kaspar-Schoenefeld,& nbsp et al.,& nbsp High throughput 4D-Proteomics – Application of dia-PASEF & reg and the Evosep One for short gradients. App Note 1867805 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Thomas Kosinski,& nbsp et al.,& nbsp Maximized throughput and analytical depth for shotgun proteomics using PASEF on a TIMS equipped QTOF. ASMS 2018, TP 685 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Thomas Kosinski,& nbsp et al.,& nbsp Plasma proteomics goes high throughput-timsTOF Pro with PASEF and 4D feature alignment to quantify 500 plasma proteins in 11.5min. App Note 1867805 /p p style=" margin-top: 10px margin-bottom: 10px line-height: 1.5em text-indent: 2em " · & nbsp Thomas Kosinski,& nbsp et al.,& nbsp Short nanoLC gradients optimize throughput on a tims equipped QTOF with PASEF, ASMS 2019, TP 514.& nbsp /p





















 我要推广仪器
我要推广仪器
 下载APP
下载APP