基于电荷检测质谱(CDMS)对AAV提取的DNA的分析揭示基因组的截断
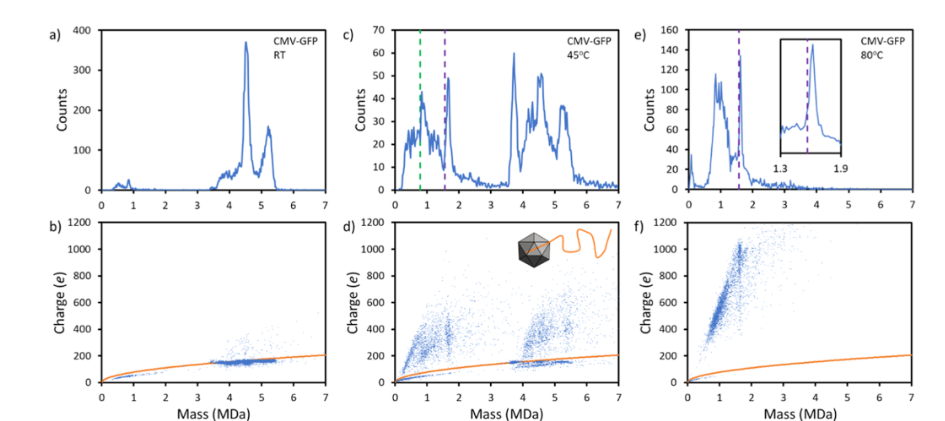
大家好,本周为大家分享一篇发表在Analytical Chemistry上的文章,Analysis of AAV-Extracted DNA by Charge Detection Mass Spectrometry Reveals Genome Truncations1,文章的通讯作者是来自印第安纳大学化学系的Jarrold, Martin F.教授。 腺相关病毒(AAV)是一种小的(26纳米)、无包膜二十面体病毒。由于其低免疫原性和高组织亲和性,AAV已成为一种很有前途的基因治疗载体。AAV衣壳包含三种病毒蛋白质,VP1、VP2和VP3。对于来自HEK细胞的重组AAV (rAAV),VP1-3的比例约为1:1:10。AAV包裹单链(ss)DNA基因组。野生型基因组的长度约为4.7 kB。基因组两侧有两个倒置末端重复序列(ITRs),它们在复制和基因组包装中起着重要作用。目前,主要用于rAAV研究的生产平台是人HEK293细胞的瞬时转染,然而其HEK293细胞的制造限制其大规模地用于AAV载体的生产。杆状病毒感染的Sf9细胞系已被发现是一种可行的生产方法,但是研究发现在生产过程中出现的ITR丢失和基因组截断现象,似乎成为了Sf9细胞系必须关注的一个问题。因为包裹着不完整的基因组的载体,会使得治疗的有效性降低。 在本研究中,作者提出了一种利用电荷检测质谱(CDMS)直接检测从AAV中提取的DNA的方法。CDMS可以使用静电线性离子阱(ELIT)同时检测单个粒子的电荷数和质荷比,从而直接获得粒子的质量。测量是在一个自制的仪器上进行的,简单地说,纳喷雾(Advion Triversa Nanomate)产生的离子通过金属毛细管进入仪器,然后通过几个不同真空区域。第一个区域包含FUNPET(an ion-funnel ion-carpet hybrid),随后是射频六极杆和分段射频四极杆。FUNPET会破坏气体通过毛细管时形成的气体射流,样品离子随即在六极杆中被热化,最终的离子能量由六极杆上的直流电位决定。离子束在分段四极杆中的径向分布被压缩,经过四极杆的离子通过非对称艾泽尔透镜聚焦到双半球形偏转能量分析器中,并设置传输具有较窄动能分布的离子(以100 eV/z为中心)。传输的离子被聚焦到ELIT中,其中一些离子被捕获并通过位于ELIT端帽之间的检测圆筒来回振荡。振荡离子产生的信号被电荷敏感放大器接收。信号被放大和数字化,然后用快速傅里叶变换(FFTs)进行分析。短时间窗口FFT通过每个捕获事件的信号进行转换,以确定离子是否在整个事件中被捕获。没有在整个事件中存活的离子信号将被丢弃。振荡频率与m/z有关,振幅与电荷成正比。用这种方法测量了数千个离子,并将其分成直方图以给出质量分布。 图1. 来自Sf9细胞的AAV8-CMV-GFP的CDMS测量。(a,b)未孵育样品的质量分布和电荷与质量散点图。电荷与质量散点图中的橙色线是球形离子瑞利电荷极限的预测。(c,d)在45°c孵育15分钟后测量的质量分布和散点图。(d)中的插图显示了基因组从衣壳挤出的示意图。(e,f) 80°C孵育15 min后的结果。绿色虚线表示释放的ssDNA GOI的序列质量,紫色虚线表示互补DNA链碱基对进入溶液后的序列质量。图1第一排的图片显示了用CDMS测量的Sf9细胞制备的AAV8-CMV-GFP的质量分布。在4.5MDa处的主峰是由于rAAV对GOI进行了包装,在5.2MDa处的峰值是由于异质DNA的包装达到了包装容量,在3.7处MDa的肩峰是由于空颗粒。对应的电荷-质量散点图如图1第二排所示。其中空颗粒和包装了DNA的颗粒在电荷上的数值比较接近是因为DNA被包裹到了衣壳的内部。图1c显示了AAV8-CMV-GFP在45°C孵育15min后测量的质量分布。rAAV已经开始分解,存在大量质量低于3 MDa的离子。在3.7 MDa处的空颗粒的数量也大幅增加,这表明基因组正在被释放。而在80℃孵育15min后可见AAV已经完全分解,对应峰也消失了,而剩下的峰与推测的互补DNA链的分子量相当。图2显示了培养后为提取GOI而测量的rAAV载体的CDMS质量分布和电荷-质量散射图。值得注意的是,AAV8-CMV-CRE和AAV8-CAG-GFP(来自Sf9细胞)的平均电荷约为400 e, AAV8-CMV-GFP(来自HEK细胞)的平均电荷约为900 e。平均电荷的差异可能反映了dsDNA的整体几何结构,电荷越高的GOIs具有更广泛的结构。 图2. 在80°C孵育15分钟后记录的代表性质量分布和电荷与质量散点图。结果显示AAV8-CMV-CRE、AAV8-CAG-GFP和AAV8-EF1a-GFP来源于Sf9细胞,AAV8-CMV-GFP来源于HEK细胞。紫色虚线显示dsDNA GOI的序列质量。插图显示了dsDNA GOI的峰值的扩展视图。图3a显示了测量到的dsDNA GOI与AAV样本序列质量的偏差的柱状图,对于大多数AAV样本,测量的dsDNA GOI大于序列质量。这种偏差可以用反离子来解释。DNA在中性溶液中带负电荷,因为它的一些主链磷酸被电离,dsDNA GOI有2219−3443个碱基对,因此它们可能有多达4438−6886个反离子。最可能的反离子是NH4+因为样品是用醋酸铵溶液电喷涂的。如果所有的dsDNA GOI主链磷酸都被电离并且有NH4+反离子,则附加质量(超出完全电离序列质量)为80 ~ 124 kDa。而有些dsDNA的分子量低于预测的序列质量,这是因为序列发生了截断导致的,图3d显示了为该样品测量的DNA峰值的扩展视图。峰宽可以提供截断分布的信息。如果所有的DNA链都损失了425 nt,峰值就会很窄。另一方面,如果截短长度分布较宽,则会产生较宽的峰值。图3d中的峰值相对较窄,说明分布较窄。有一个高质量拖尾,这可能表明一些基因组被截断了小于425 nt。 图3. 来自Sf9和HEK细胞的一系列GOIs的AAV8、AAV9和AAVDJ血清型的dsDNA质量测量总结。(a)测量质量与序列质量偏差的柱状图。(b)考虑反离子的测量质量与预期质量的偏差的柱状图。(c) AAV基因组结构示意图。(d)来自HEK细胞的AAV8-CMV-CRE的dsDNA GOI峰的扩展视图。最后,将CDMS测量的基因组截断与来自第三代测序方法的信息进行比较将具有指导意义。尽管CDMS测量可以判断基因组是否被截断以及缺失的数量,但它不能确定截断发生在哪里。关于截断发生位置的信息可以从第三代测序中获得,这些信息反过来可以深入了解其机制。因此,CDMS测量全基因组MW和第三代测序是互补的。CDMS测量可用于筛选截断的基因组,以便通过第三代测序进行后续深入分析。 撰稿:李孟效 编辑:李惠琳 文章引用:Analysis of AAV-Extracted DNA by Charge Detection Mass Spectrometry Reveals Genome Truncations 李惠琳课题组网址www.x-mol.com/groups/li_huilin 参考文献 1. Barnes, L. F. Draper, B. E. Kurian, J. Chen, Y. T. Shapkina, T. Powers, T. W. Jarrold, M. F., Analysis of AAV-Extracted DNA by Charge Detection Mass Spectrometry Reveals Genome Truncations. Analytical Chemistry, 4310-4316.


















 我要推广仪器
我要推广仪器
 下载APP
下载APP