李惠琳团队成果:非变性自上而下质谱用于蛋白及其复合物结构表征
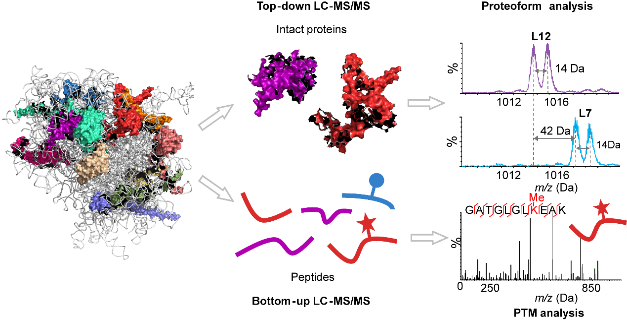
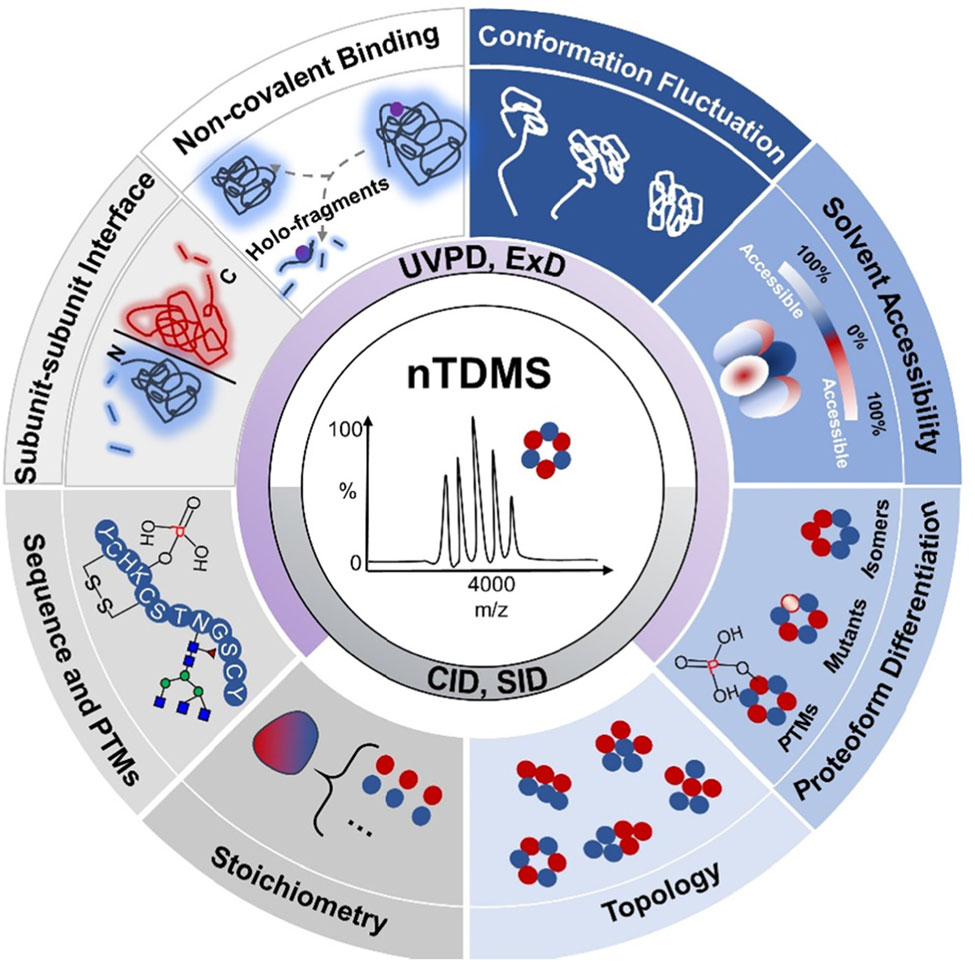
大家好,本周为大家分享一篇李惠琳课题组最近发表在Mass Spectrometry Reviews上的综述,Native top‐down mass spectrometry for higher‐order structural characterization of proteins and complexes1。结构生物学的快速发展极大地促进了蛋白结构表征工具的开发。其中,基于质谱的分析方法凭借其快速、灵敏、高通量的优势从中脱颖而出。相比于原子水平的高分辨结构表征工具如X-射线晶体学、核磁共振(NMR)、冷冻电镜(Cryo-EM)等,基于质谱的分析方法能够有效地补充蛋白动力学结构变化的信息,并且不受蛋白纯度、分子量大小的限制。而相较于低分辨的蛋白表征工具如圆二色光谱、动态光散射等,基于质谱的分析方法能够提供更高的肽段或残基水平分辨率,获取额外的序列、翻译后修饰(post‐translational modifications, PTMs)、局部空间结构等信息。常见的结构质谱包括:氢氘交换质谱(hydrogen‐deuterium exchange MS, HDX-MS)、交联质谱(cross‐linking MS, CX-MS)、表面标记质谱(covalent labeling MS, CL-MS)等。已有相当多的文献对这些方法进行了详细的介绍2,3,在此不再赘述。而此篇综述将重点介绍非变性至上而下质谱(native top‐down MS, nTDMS)在蛋白及其复合物结构表征中的应用。在过去的十年,非变性质谱(native MS, nMS)特别是nTDMS发展迅速。nMS作为一个桥梁将蛋白质组学与结构生物学相连,其保留非共价相互作用的特性使其广泛用于蛋白复合物四级结构表征,如推断亚基组成、化学计量比、亚基排布等。然而,对于一些深层次的结构信息,如氨基酸序列、PTMs、配体结合位点、亚基结合界面等,仅靠单一的nMS是无法获取的。与之对应的,变性条件下的自上而下质谱(TDMS)能够在完整蛋白水平下直接获得序列以及PTMs信息,虽然有助于PTM的准确定位以及蛋白、蛋白异质体(Proteoform)的鉴别,但却丢失了涉及非共价相互作用的高级结构信息。受限于质谱仪器的发展,在早期,nMS与TDMS通常在两个独立的实验中进行,随着质量分析器以及多种活化/碎裂方式的开发,nMS与TDMS的能够有效的结合,充分发挥各自的优势,在实现多层次结构信息获取的同时,也在不断挑战更加复杂的生物体系,如核糖体、膜蛋白、内源蛋白混合物等。实验设计nTDMS已成为表征蛋白质和复合物的初级到高级结构的重要工具。随着蛋白质样品的大小和复杂性的增加,用于nTDMS的仪器不仅需要符合某些特定标准,还需要不断提高其性能以满足这些增加的需求。nTDMS分析中几个关键的步骤包括:样品前处理、ESI离子化、二级碎裂、质量检测以及数据处理。样品前处理为了维持蛋白的自然状态,通常需要在生理环境中进行nMS分析。然而,缓冲液中的非挥发性盐会产生大量盐簇并与蛋白离子形成非特异性加合物,从而抑制离子信号、降低检测的准确度和灵敏度。因此,样品前处理过程中最重要的环节就是除盐。然而适当的离子强度有助于维持蛋白的三维结构,所以通常的步骤是对蛋白进行缓冲液置换,将蛋白置换至醋酸铵或碳酸氢铵等挥发性盐溶液中。目前已开发了多种在线或离线的除盐方法,详细内容的可在综述原文中查看,此处不再赘述。除了使用非挥发性缓冲盐,减小ESI喷针孔径大小也可以提高系统耐盐能力。碎裂/活化方式二级碎裂方式是实现nMS到nTDMS的关键。常见的活化方式按照原理可分为三类:基于碰撞(CID, SID)、基于电子(ECD, ETD, EID等)以及基于光子(UVPD, IRMPD)的活化/碎裂方式。值得注意的是,CID与IRMPD都属于慢加热的活化方式,能量累积的非常慢,以至于在发生碎裂之前已经进行了能量重排,一些较弱的或者不稳定的键会优先发生断裂,最终导致非共价相互作用在活化的过程中被破坏。而SID、ExD与UVPD则属于快加热的活化方式,碎裂发生在能量重排之前,非共价相互作用得以在这一过程保留下来,碎片化程度受到非共价相互作用的限制,因此可被用于表征蛋白的空间结构。此外,将多种活化方式的结合或与离子淌度技术串联也是获取多层次结构信息的关键。质量检测与变性条件下的质谱分析相比,蛋白复合物在天然环境下通过电喷雾电离产生的电荷数相对较少,因此需要具有较大m/z 范围的质量分析仪(高达m/z = 20,000 Da甚至更高)。最初,nMS分析高度依赖基于飞行时间(time of fight, TOF)质量分析器,因为TOF具有理论上无限的m/z范围。近年来,高分辨质量分析器如轨道阱(Orbitrap)和傅里叶变换离子回旋共振(FTICR)为生物大分子的nTDMS分析带来了新的活力。在综述中,我们简要介绍了每种质量分析器的最新进展,并重点强调了FTICR和Orbitrap在nTDMS分析中的发展和应用。数据处理除了基本的硬件设施,配套的数据处理软件也十分重要。nTDMS数据处理流程通常包括以下4个步骤:同位素峰选取、去卷积、数据库搜索、验证和可视化。正文中,我们对每个步骤进行了简要描述,并重点介绍用于数据库搜索和异质体鉴别的软件。多层次结构信息的获取得益于多种活化/碎裂方式的开发,nTDMS分析可同时获得多层次的结构信息(图1)。主要有以下两种策略:第一种策略,完整蛋白复物(MS1)首先被CID或SID碎裂至亚基(MS2),亚基可进一步碎裂肽段(MS3),在MS1及MS2中可获蛋白复合物结合计量比、拓扑结构、蛋白异质性等信息,在MS3阶段则可获取蛋白序列、PTMs定位以及异质性来源等信息。第二种策略则是完整蛋白复合物(MS1)直接被UVPD或ExD碎裂成肽段(MS2),受益于UVPD以及ExD独特的碎裂方式,发生碎裂的区域主要位于蛋白复合物的表面可及区,而未发生碎裂的区域可能位于蛋白复合物的核心区域或参与亚基相互作用界面。不同的碎裂情况反映不同的空间结构,带有配体的肽段碎片可以用于配体结合位点的定位。综述中,我们详细阐述了如何利用nTDMS获得蛋白复合物的多层次结构信息以及如何将碎片信息与结构信息相关联。图1. nTDMS可提供的多维度结构信息复杂生物体系中的应用蛋白质的空间结构决定了其生物功能,而蛋白质-蛋白质/配体相互作用是大多数生物进程的基础。通过突变、翻译后修饰、或者与金属、小分子配体、蛋白质、DNA、RNA等分子发生共价或非共价的相互作用,蛋白质功能在活细胞中不断受到调节。随着MS仪器、方法的不断开发和数据处理软件的逐渐成熟,nTDMS已被广泛应用于各种生物系统,从小蛋白质、蛋白质-配体复合物到大分子组装体,如膜蛋白、蛋白酶体、核糖体、病毒衣壳,甚至是内源性蛋白混合物。它们中的许多都是极具挑战性的体系,即便是采用NMR、X-射线晶体学或Cryo-EM等生物物理方法分析也是非常困难的。因此,来自nTDMS的见解对于理解这些蛋白质和复合物至关重要。在这里,我们总结nTDMS在所有生物体系中的应用实例,旨在全面了解nTDMS在解决生物学问题方面的潜力。小蛋白的结构表征和区分最初,nTDMS主要用于50 kDa以下单体蛋白的结构表征,大部分的研究都是围绕蛋白质气相结构与溶液相结构对比展开的。根据nTDMS的碎裂情况,推断蛋白的气相空间结构,并与NRM获得的溶液结构进行对比。此外,如果在二级碎裂前增加离子预活化有助于蛋白分子的展开,以便研究蛋白气相展开路径以及获取蛋白质内部空间结构信息。得益于碎片离子对蛋白空间结构的高度敏感性,nTDMS还被用于区分不同蛋白亚型、蛋白突变体的结构差异。蛋白-小分子配体相互作用随后,nTDMS应用到了蛋白-配体复合物中,不同的配体类型适合不同的活化/碎裂方式,除了金属离子、RNA/DNA等以静电作用为主的蛋白配体能够在CID活化时存活,大部分复合物的碎裂都需要选择ECD或UVPD等方式。nTDMS可用于蛋白-配体结合计量比、亲和力、结合位点、作用机制、结构动力学/变构效应的研究。它是一种强大的结构表征工具,其在抑制剂筛选、酶催化监控、RNA-蛋白质互作机制的应用实例在正文中已有详细的介绍。蛋白-蛋白相互作用随着仪器设备的快速发展,nTDMS已应用到更大的体系如蛋白-蛋白复合物,通过组合不同的活化/碎片化技术,在一次实验中可以获得多层次的结构信息。nTDMS可以帮助区分不同的蛋白异质体,并在完整复合物、亚基、肽段三个水平上确定异质性的来源。蛋白的异质性与其生物学功能密切相关,通过调整蛋白的异质性可以实现蛋白功能的转变,具体的应用案例已在正文详细介绍。除此之外,nTDMS还可以用作蛋白-蛋白复合物结合界面、气相展开以及深层次结构探索。治疗性抗体和抗原-抗体复合物在过去的几十年中,治疗性抗体已成为最受欢迎的候选药物之一,它们的高特异性和低副作用促进了治疗性抗体的快速增长。在综述中,我们还详细地介绍了nTDMS在治疗性抗体和抗原-抗体复合物体系中的应用。nTDMS可用于抗体可变区的测序、具有不同药物计量比(DARs)的抗体耦联药物的结构表征、以及抗体-抗原复合物中互补决定区及抗原表位区的鉴别。膜蛋白无论是对于传统的结构表征工具如:X-射线晶体学、NMR还是nTDMS,膜蛋白的结构表征一直以来面临着诸多困难。膜蛋白具有低丰度以及低溶解性等特点,最常见的方法是利用与nMS兼容的膜模拟物如:去污剂胶束、纳米微盘等去溶解膜蛋白,在nTDMS分析时再将膜蛋白从胶束中释放出来,释放出的蛋白可在nTDMS中进一步碎裂获取结构信息。具体的实验流程和应用实例可在综述正文中查看。大分子组装体正文中,还介绍nTDMS在极具挑战性的大分子组装体如:核糖体、蛋白酶体、病毒衣壳中的应用实例,这些生物体系普遍存在的问题是分子量非常大(接近MDa),且具有较高的异质性。对这些大分子机器进行nTDMS分析要求仪器具有较高的质量范围以及分辨率。大分子机器的结构表征充分说明nTDMS方法无论在深度还是广度上都有极大的提升。Native top-down MS蛋白质组学值得注意的是,当质谱前端结合非变性分离技术,如native GELFrEE,尺寸排阻色谱,毛细管区带电泳,离子交换色谱等,nTDMS还可以在靶向模式或发现模式下用于复杂蛋白质组的高通量分析,如内源性蛋白混合物。nTDMS分析最大的优势在于它能区分不同的蛋白异质体,并对每种蛋白异质体进行结构表征,这是其他在肽段水平进行分析的结构质谱法如:HDX-MS, CL-MS所无法实现的。总结与展望总之,在这篇综述中我们重点介绍了nTDMS的最新进展和在不同生物体系中的应用,强调通过nMS与TDMS结合可以获得额外的多层次结构信息。新技术的出现以及仪器的进步使nTDMS能够应用于结构生物学中日益复杂的生物样本体系,包括蛋白质配体、多聚蛋白复合物、大分子组装体和内源性复合物。尽管这样,nTDMS分析仍面临着的挑战,包括但不限于前端的样品分离、离子化、去溶剂化、高质荷比分子传输、异质性样本的分析以及软件的开发。未来nTDMS将与其他的一些结构表征方法相结合以获取更加全面的结构信息。正文中对未来发展趋势进行了讨论并提到了其他一些令人兴奋的创新技术如:基于MALDI离子源的质谱成像技术用于蛋白原位分析、电荷检测质谱(CDMS)用于异质性样本分析,多重技术的结合将为蛋白质复合物的nTDMS研究开辟新的道路。我们希望这篇综述能让读者更好地理解nTDMS提供的独特结构信息,并推动该方法的广泛应用。撰稿:刘蕊洁编辑:李惠琳原文:Native top‐down mass spectrometry for higher‐order structural characterization of proteins and complexes. 参考文献1.Liu RJ, Xia SJ, Li HL. Native top‐down mass spectrometry for higher‐order structural characterization of proteins and complexes. Mass Spec Rev. 2022 e21793. https://doi.org/10.1002/mas.217932.Britt HM, Cragnolini T, Thalassinos K. Integration of mass spectrometry data for structural biology. Chem Rev. 2022 122(8):7952-7986. 3.Liu XR, Zhang MM, Gross ML. Mass spectrometry-based protein footprinting for higher-order structure analysis: fundamentals and applications. Chem Rev. 2020 120(10):4355-4454.


















 我要推广仪器
我要推广仪器
 下载APP
下载APP