药品评价和研究中心(Center for Drug Evaluation and Research [CDER])向美国人民承诺可用上安全和有效的药品。在《1906年食品和药品法》(1906 Pure Food and Drugs Act)出台前,该中心仅由个人操作且只对市场上重大药品问题进行评价,至今已在职责和组织上发生了变化。这种变化部分地反映了药品法的演变和20世纪化学疗法的革新——以及FDA的职责的变化。但这种变化也反映了对于如何更好地向病人提供安全有效的药品的外部和内部的决定。政府的各个部门,和一些利益受FDA政策所影响的部门,对FDA的药品管理发挥了一定的作用。以下简述在过去的90年中FDA药品管理职能的变化,尤其是组织上的剧变。 在1902年的美国药物协会(American Pharmaceutical Association)年会上,化学局(Bureau of Chemistry)的首席药剂师(Chief Chemist)Harvey Wiley,宣布在其下面成立一个药品实验室(Drug Laboratory)。Wiley希望这个实验室会有助于药物标准化并统一分析结果。Lyman Frederic Kebler,是法国Smith Kline的首席药剂师,以及公认的假药鉴别专家,被任命为该实验室的领导者之一。他于1902年12月被任命为药品实验室主管,于1903年三月上任。 起初,药品实验室从事各种不同的项目。其一是对该局所用试剂的调查,很快Kebler发现试剂不纯。他还花大量时间寻求改进药物分析的方法。Kebler还提醒公众关注通常的药品供应中存在的问题。 三年后,在Wiley的长期游说下,《食品和药品法案》成为法律。它禁止冒牌的和掺假的药品和食品(mislabeled and adulterated drugs and food)在州际贸易。Wiley自身强调食品是关系健康的一个重要因素,但他同时也对专利药(patent medicines)和处方药(prescription drugs)给予了一定的关注。他的继任者——Carl Alsberg,提高了药物的重要性。1908年,药品实验室经历了第一次重要的重组,更名为药品处(Drug Division),下设四个实验室:1、药品检查实验室(Drug Inspection Laboratory),由George Hoover主管;2、合成成品实验室(Synthetic Products Laboratory),由W. O. Emery 主管;3、提炼油实验室(Essential Oils Laboratory),由E. K. Nelson主管;4、药理学实验室(Pharmacological Laboratory),由William Salant 主管。Kebler仍是处长。 提炼油实验室是对油类进行分析检测,无论是单独用于治疗还是与其它化合物结合在一起的,如Root-beer提炼物,和鹿蹄草(wintergreen)提炼油;合成实验室负责对合成药物的检查,包括普通头痛药物,以及原料药中的活性成分;药理学实验室负责调查药品对动物的生理影响,主要是针对咖啡因,这是Wiley很感兴趣的一个课题;药品检查实验室是该处的一个主要执行部门,比如,从1909—1910年,这个实验室仔细考察了900多个国内药品的样品,大约1000个进口样品,并检举了115个样品。 在1910年,根据1906年的法案管理药品时,出现了较大的冲突,当局查获了大量的“Johnson的温和治癌药”(Johnson's Mild Combination Treatment for Cancer),其实是一种无用的产品,但其标签上对其疗效作了虚假的声明。审讯时,法官判定这种虚假声明不在食品和药品法管理范围之内,并作出了不利于政府的裁定。1912年,国会颁布了修正后的法律。《Sherley修正案》(Sherley Amendment)中将疗效声明归入食品和药品法的管理权限内,但在判定其违法之前,要求该局出示其声明是错误的或是虚假的证明。 到这时,药品检查实验室的工作范围增加了。比如,他们不仅对检测吗啡、硝化甘油的方法进行调查,还同美国药典会(U.S. Pharmacopeia[USP])一起研究药品标准。药品处的工作不仅局限于国内药品,还针对进口药品、化合物以及进口的疗效可疑的产品。该处室还花大量时间调查受污染的氯仿。一些制造商用马口铁容器装氯仿,使其容易分解,而USP规定氯仿必须贮存在玻璃容器中。 1910年代中期,药品处新增了两个部门。其中一个是生药学实验室(Pharmacognosy Laboratory),创立于1914年。它除了调查原料药成品以外,还研究如何改进原料药工艺以减少浪费。1916年在Sherley修正案的影响下,又成立了一个办公室专门调查药品的错误和虚假标识(false and fraudulent labeling of drugs),由M.W.Glover主管,他是美国公众健康服务部(U. S. Public Health Service[USPHS])的一位内科医师。 1920年该局成立了独立的药品管理办公室(Office of Drug Administration),由Glover主管,协助药品处管理药品标签的发行细节。1923年由于Glover被召回USPHS,这个办公室就取消了。同时,成立了一个自主的特别合作调查办公室(Office of Special Collaborative Investigations),由Kebler主管,与邮政部(Post Office Department)一起调查有假发行物的邮件,药品处也改组了。1923年,药品控制办公室(Office of Drug Control)代替了药品管理办公室和药品处,由George Hoover主管,它在组织上与新成立的食品控制办(Office of Food Control)保持平行,它对药品(包括原料药、药品组分、药物制剂和专利药品)控制中的所有工作负责。 药品控制办继续与相关方保持合作,如贸易协会和USP。提高制造业正确度的愿望使协会间形成了联系委员会。当委员会完成提高正确度的方法的研究后,在商业期刊上就会刊登出来。1926年,该局与USP合作通过一个共同的项目。该项目是为了提供生物分析药品的标准化产品样品,例如洋地黄(digitalis)作为对照品的研究,该项目一直持续到1930年。 接到涉及不纯麻醉剂导致几起死亡的报告后,该办着手调查原因。当时市场上有好几种麻醉剂,但他们主要是对乙醚和乙烯的调查。该调查一直持续到1930年代中期。通过调查分析,药品控制办确定麻醉剂的分解是由于制造工艺较差引起的。作为调查的一部分,药品控制办设立了一个实验室专门致力于药品分析。 1928年起,药品、食品和杀虫剂管理局(Food, Drug and Insecticide Administration)下的药品管理部门,经历了几次重大的人事变动。George Hoover在主管药品控制办公室5年后离开了该局,Lyman Kebler被重新任命为特别合作调查办主任。从而特别合作调查办就成为药品控制办下面的一个组,此时它下面已有一个化学小组(Chemical Unit)、医学小组(Medical Unit)、兽药小组(Veterinary Unit)和药理学小组(Pharmacology Unit)。新主管是James J.Durrett,他与Kebler一样是一个内科医师和药师;当时Durrett是Tennessee大学的公众健康学教授。 由Marvin R.Thompson 指导的对麦角碱(ergot)药理的研究是该办的一项重要研究工作。起初,他们注意的是麦角碱的分解;分析表明这主要出现在运送过程中。但在这个调查中,对麦角碱药理缺少了解成了突出的问题。为此Thompson开始了他的工作。在1929年,由于他的论文在药理方面有突出贡献,从而获得了美国药物协会的Ebert奖。Tompson的论文在很多领域具有突破性进展。他建议修改麦角碱的分析方法,且表明通过改变麦角碱 的制备工艺可以改进USP规定的麦角碱标准。 自从《食品和药品法案》成为法律后,它的缺点变得明显了。其中一个缺点是无权阻止声称减肥的危险的制剂的销售。1934年,药品控制办(从1931年以来一直由Frederick J.Cullen主管)开始调查含二硝基酚(dinitrophenol)的产品。这是饮食处方中的一个成分,能使新陈代谢率上升到危险水平,并引起许多死亡和伤害。由于该法律没有对药品安全作出规定,因此,药品控制处无权没收产品,只能提出警告。 1935年,Jame J. Durrett回到FDA(Food and Drug Administration)主管药品管理部门,该部门再次被命名为药品处(Drug Division)。同年,以前一直属于药品处的药理学实验室成了一个独立的办公室,由Erwin E. Nelson主管。Nelson是作为该办的唯一的顾问,而同时从1919—1937年又是Michigan大学的教员,从1937—1943年是Tulane大学药理系的主任。该办公室的独立是为了适应食品工业中不断上升的对药理学方面的需求。尤其是在调查食品中的有毒物质和杂质的时候。
作者 华丛笑 高晨燕 正文内容 摘要:美国食品药品监督管理局已经走过了百年历程,建立了较为完善的药品监管体系。本文通过比较中美两国的药品说明书管理现状,为我国完善和发展药品说明书的管理提供启发和借鉴。关键词:中国,美国,药品说明书,管理药品说明书的信息直接关系到安全、合理使用药品。药品说明书的评价研究表明,很多药品不良反应的主要原因是说明书的信息不清楚。此外,药品说明书具有法律效力,在发生药品使用相关的医疗纠纷时,能够为之提供法律依据。因此,药品说明书的规范化已经成为我国药品管理的现实要求。美国FDA在药品说明书的监管方面积累了大量成熟的经验,本文旨在通过比较中国和美国化学药品说明书的管理现状,引起国内药政管理人员、医药专家和患者的共同关注。 药品说明书管理的法律法规支持体系 我国对药品标签和说明书管理的法律法规支持体系主要有法律、行政法规、以及SFDA发布的具体细则和规定等。 法律和法规:①《中华人民共和国药品管理法》(1984.9.20全国人民代表大会通过和颁布,2001.2.28修订),第54条规定,药品包装必须按照规定印有或者贴有标签并附有说明书。②《中华人民共和国药品管理法实施条例》(国务院颁布),第46条是关于药品标签和说明书的规定。③《药品注册管理办法》(SFDA颁布),其中第十一章的第三节(第一百四十二条-第一百四十五条)规定了药品说明书和标签的责任者、审核者和核准机构。 为了规范药品说明书和标签的格式和内容,SFDA先后发布了《药品包装、标签和说明书管理规定(暂行)》、《药品说明书规范细则》和《药品说明书和标签管理规定》等。此外,2007年10月,国家食品药品监督管理局发布含有兴奋剂的药品目录,要求其在药品说明书或者标签上注明“运动员慎用”字样。 美国与药品有关的法律文件大致分为3类[1]:国会批准的法律、联邦政府各行政部门颁发的联邦法规和FDA制定的指导原则。法律和联邦法规是企业必须遵守的法律要求,具有法律强制性;而指导原则属于建议性质,供企业参考使用。按照美国法规的规定:所有的药品都必须有充分的标签说明(这里的标签包括药品说明书、标签和其他所有附加于药品的书写物、印刷物、绘制物)。 美国与药品说明书和标签相关的法律包括:1906年《清洁食品和药品法》(Pure Food and Drugs Act,PFDA);1938年《联邦食品药品化妆品法》(Federal Food, Drug,and Cosmetic Act, FDCA) ; 1951年《Durham-Humphrey修正案》(Durham-HumphreyAmendments);1962年《Kefauver-Harris药品修正案》(Kefauver-Harris Drug Amendments);1966年《正确包装和标志法》(Fair Pack-aging and Labeling Act,FPLA);1988年《处方药市场营销法》(Prescription Drug Marketing Act);1997年《食品和药品管理现代化法》(Food and Drug Administration Modernization Act,FDAMA)。联邦法规主要是指美国《联邦法典》(CFR),药品部分主要集中第21篇(简写为21CFR),药品标签和说明书的相关法规收编于21CFR.210和330。 FDA发布与药品说明书管理配套的指导原则,使法规的要求得以深入和具体化,使企业在撰写说明书时有据可依。例如:FDA发布《人用处方药品及生物制品说明书格式及内容管理条例》之后,为了指导申请人及生产企业执行新的管理要求,帮助已批准药品说明书的修订完善及新旧版式转换,制定了一系列配套的指导原则,包括“药品说明书新内容格式管理条例实施指导原则”、“药品说明书[临床研究]内容格式指导原则”、“药品说明书[不良反应]内容格式指导原则”、“药品说明书[注意事项]、[禁忌症]、[黑框警告]内容格式指导原则”等。 就药品说明书管理的法律法规支持体系而言,美国已经建立起一个庞大的法律框架支持药品说明书的管理,使企业在撰写说明书时有据可依,药品监管部门对药品说明书的审核具有独立性。而我国的药品监管历史始于近20多年,药品说明书管理相关的法规和指导原则尚有待不断细化和完善,尚未建立独立的药品说明书的审核体系,还需要参照FDA或/和EMEA的相关内容作为重要的参考依据。 药品说明书的管理机构 我国药品说明书的管理机构 《药品注册管理办法》规定,药品说明书和标签由申请人提出,由国家食品药品监督管理局核准。 我国具体负责审核药品说明书的机构包括:SFDA及其下属的药品审评中心、药品评价中心和省级食品药品监督管理部门。根据说明书的审核内容的不同而进行分级负责管理。例如:新上市药品的说明书、已经上市的药品增加适应症由国家药品审评中心审核、国家食品药品监督管理局注册司批准。而OTC药品的说明书由SFDA下属的药品评价中心审核。根据SFDA的要求修改进口药品说明书;补充完善进口药品说明书的安全性内容等则由省级食品药品监督管理部门批准。根据国家药品标准或国家食品药品监督管理局的要求修改国内生产药品说明书;补充完善国内生产药品说明书安全性内容等直接向省级食品药品监督管理部门备案。 美国药品说明书的管理机构 FDA下属的药品评审与研究中心(Center for Drug Evaluation and Review,CDER)负责药品说明书和标签的审核,监督所提供的药品信息的真实性和准确性。说明书通常是先由企业起草,并且向FDA提供支持说明书各项内容的研究数据,FDA对该说明书进行审核,并且和企业做进一步的讨论,形成终稿。FDA批准的说明书通常和批准信一起放在FDA的网站上,此外在National Library of Medicine’s Daily Med的网站上也可以找到FDA核准的电子说明书。 两国审核和核准药品说明书的职能部门分别为FDA和SFDA以及它们下属的机构。在中国,SFDA及其下属的药品审评中心、药品评价中心和省级食品药品监督管理部门均根据职责规定对说明书进行形式与内容的审核管理,然而尚存在的一个问题是:目前上市流通的同一品种的说明书尚存在少数具体内容的差异现象。 药品说明书的形式和内容管理 两国对药品说明书的形式和内容管理的目的均是最大程度地确保用药安全和有效。 我国2006.6.1颁布的《药品说明书和标签管理规定》标志着我国药品说明书管理的重要举措,规定了其格式和内容的具体要求:要求药品生产企业应在药品说明书中完整、规范地标明相关的项目内容;强调说明书必须提供准确、清晰、详细而全面的药品信息。例如:必须注明药品的全部活性成分或中药药味,强调药品不良反应信息的充分说明(列出所有已知的不良反应);必要时须加注警示语等。此外,SFDA颁布相关的指导原则和说明书范本供企业参考,例如《化学药品、生物制品说明书指导原则》、《中药、天然药物药品说明书撰写指导原则》、《体外诊断试剂说明书指编写指导原则》、OTC化学药品说明书范本以及OTC中药说明书范本。
无菌药品技术转让审批权重新收回总局 从国家食品药品监督管理总局(CFDA)获悉,2015年1月1日起,注射剂等无菌药品技术转让的审评审批权将收回到总局,此前已获总局批复授权的省级药品监管机构也将停止该项工作。 CFDA表示,自2015年1月1日起,注射剂等无菌药品的技术转让注册申请,应当按照《药品技术转让注册管理规定》的程序和要求申报补充申请,由国家食品药品监督管理总局进行审评审批;已获总局批复授权的省级药品监管机构,停止受理注射剂等无菌药品的技术转让注册申请;已经受理的注册申请,要认真审查工艺验证、质量对比等研究资料,不符合要求的,坚决不予审批。 2013年2月,CFDA发布《关于做好实施新修订药品生产质量管理规范过程中药品技术转让有关事项的通知》,对于在药品GMP改造过程中药企整体搬迁、兼并等情形涉及的药品技术转让问题作出规定,并授权省级药监部门管理。其中,注射剂等无菌药品生产企业应在2014年12月31日前提出转让注册申请。此次CFDA是在该时间节点之后,将权力收回到总局。
药学研究:中检所 说明:中检所 --[科研中国 SciEi.com]网址:http://www.nicpbp.org.cn 药品审评中心 说明:药品审评中心 --[科研中国 SciEi.com]网址:http://www.cde.org.cn/ 药典委员会 说明: --[科研中国 SciEi.com]网址:http://www.chp.org.cn/ 药品认证管理中心 说明: --[科研中国 SciEi.com]网址:http://www.ccd.org.cn/ 国家中药品种保护审评委员会 说明: --[科研中国 SciEi.com]网址:http://www.zybh.gov.cn/ 药品评价中心 说明: --[科研中国 SciEi.com]网址:http://www.cdr.gov.cn/ 医药出版社 说明: --[科研中国 SciEi.com]网址:http://www.emedicity.com.cn/ 中国医药国际交流中心 说明: --[科研中国 SciEi.com]网址:http://www.ccpie.org/ [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=74669]药品申报[/url]
美国食品药品管理局(Food and Drug Admistraton简称FDA),隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化妆品、兽药、医疗器械以及诊断用品等的管理。FDA下设药品局、食品局、兽药局、放射卫生局、生物制品局、医疗器械及诊断用品局和国家毒理研究中心、区域工作管理机构,即6个局(有的刊物也称6个中心),一个中心和一个区域管理机构。美国食品药品管理机构共有职工约7500人, FDA总部有 1143人,其中药品局为350人。 药品局(也称药品评价和研究中心)负责人用药品审批工作,设有8个处和若干科室。1.药品管理处。下设药品信息、信息系统设计、行政管理和预算、医学图书馆4个科室。2.药品监督办公室。下设有药品质量评价、药品标签监督、生产和产品质量、科研调查、法规等7个科室。3.药品标准处。设有常用药品评价、药品上市和广告2个科。4.药品审评一处。下设心血管--肾脏药、抗肿瘤药、营养药、医用造影外科和齿科药、肠胃药和凝血药5个科室。5.药品审评二处。下设抗感染药、代谢和内分泌药、抗病毒药3个科室。6.流行病和生物统计处。下设流行病及调查、生物统计2个科室。7.研究处。下设研究和测试、药物分析2个科室。8.仿制药品处。下设仿制药品、生物等效2个科室。 美国食品药品管理局设在华盛顿特区及马利兰州罗克威尔城,机构庞大,分支机构遍布全国各地。为了加强药品质量管理,FDA将全国划分成6个大区,即太平洋区(旧金山、西雅图、洛杉肌)、西南区(达拉斯、丹佛、堪萨斯)、中西区(芝加哥、明尼阿波利斯、底特律)、东北区(波士顿、纽约、布法罗)、中大西洋区(费城、辛辛那提、纽瓦克、巴尔的摩)、东南区(亚特兰大、纳什维尔、新奥尔良、奥兰多、波多利各的圣吉安)。每区设立一个大区所,大区所下又设若干个地区所。太平洋区的大区所所在地为旧金山,西南区的大区所所在地为达拉斯,中西区的大区所所在地为芝加哥,东北区的大区所所在地为波士顿,中大西洋区的大区所所在地为费城,东南区的大区所所在地为亚特兰大。 区所负责对本地区的食品、药品、化妆品、器械、血库等进行监督检查工作。各地区所按工作需要又设立若干工作站,以保证工作面能覆盖本区范围。全美目前共有143个工作站。大区所、地区所及工作站均属FDA的各级直属机构。区所的规模视工作量而定,全美的药品 65%以上在中大西洋区生产,故该区的力量较强,共有职工 525名,其中监督员 250名,约占FDA总部监督员的1/4,分析检验人员150名。各州对药品的管理按地方药品管理法规进行,主要工作是:对药师进行考试和注册、对药品经营部门和药房进行监督检查,发放或换发许可证、吊销违法户的许可证、对所在地的药学院校进行评价、审查见习药房等。
[b] 7日,国家药监局局长邵明立表明,为确保药品审评审批的公开透明,今后将会使整个过程网络化,接受申报者及社会各界的监督,并将审评人员公示,接受社会监督。邵明立同时透露,《药品注册管理办法》修订稿可能于10日上网公布。 [/b] 审评人员实行集体负责制 邵明立将这一公开透明的药品审评审批的办法归纳为“三制”,就是审评人员集体负责制,防止个别人滥用权力;实行责任追究制,强化对违法审批行为的责任追究;实行审评审批检验人员公示制,将药品审评审批置于社会监督之下。 邵明立说,国家的食品药品监管工作正在发生变化,同时自身队伍建设也暴露出一些问题,但药监局一直在努力,本来打算今年年中结束的食品药品专项整顿工作,将一直延续到年底。今年在年初,国家药监局已经委托各省药监局向当地的人大代表和政协委员汇报过专项整顿工作。 药管新规可能10日公示 昨日,邵明立看过本报《六问药品注册“漏洞”》一稿后表示,《药品注册管理办法》的修订工作正在进行中,可能于10日在网上公示。 据了解,现行的《药品注册管理办法》对涉及专利的药品注册工作作了一些规定,出发点是为了维护专利权人的利益,但由于药监局不掌握专利信息,难以判定专利是否侵权,对药品注册的正常工作形成了一定的干扰。 对此,邵明立表示,在新的办法里会考虑采取一定的措施,也会对鼓励创新药加以考虑。
(一)美国药政管理机构 美国食品药品管理局(Food and Drug Admistraton简称 FDA),隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化妆品、兽药、医疗器械以及诊断用品等的管理。FDA下设药品局、食品局、兽药局、放射卫生局、生物制品局、医疗器械及诊断用品局和国家毒理研究中心、区域工作管理机构,即6个局(有的刊物也称6个中心),一个中心和一个区域管理机构。美国食品药品管理机构共有职工约 7500人, FDA总部有 1143人,其中药品局为350人。 药品局(也称药品评价和研究中心)负责人用药品审批工作,设有8个处和若干科室。1.药品管理处。下设药品信息、信息系统设计、行政管理和预算、医学图书馆4个科室。2.药品监督办公室。下设有药品质量评价、药品标签监督、生产和产品质量、科研调查、法规等7个科室。3.药品标准处。设有常用药品评价、药品上市和广告2个科。4.药品审评一处。下设心血管——肾脏药、抗肿瘤药、营养药、医用造影外科和齿科药、肠胃药和凝血药5个科室。5.药品审评二处。下设抗感染药、代谢和内分泌药、抗病毒药3个科室。6.流行病和生物统计处。下设流行病及调查、生物统计2个科室。7.研究处。下设研究和测试、药物分析2个科室。8.仿制药品处。下设仿制药品、生物等效2个科室。 美国食品药品管理局设在华盛顿特区及马利兰州罗克威尔城,机构庞大,分支机构遍布全国各地。为了加强药品质量管理,FDA将全国划分成6个大区,即太平洋区(旧金山、西雅图、洛杉肌)、西南区(达拉斯、丹佛、堪萨斯)、中西区(芝加哥、明尼阿波利斯、底特律)、东北区(波士顿、纽约、布法罗)、中大西洋区(费城、辛辛那提、纽瓦克、巴尔的摩)、东南区(亚特兰大、纳什维尔、新奥尔良、奥兰多、波多利各的圣吉安)。每区设立一个大区所,大区所下又设若干个地区所。太平洋区的大区所所在地为旧金山,西南区的大区所所在地为达拉斯,中西区的大区所所在地为芝加哥,东北区的大区所所在地为波士顿,中大西洋区的大区所所在地为费城,东南区的大区所所在地为亚特兰大。 区所负责对本地区的食品、药品、化妆品、器械、血库等进行监督检查工作。各地区所按工作需要又设立若干工作站,以保证工作面能覆盖本区范围。全美目前共有143个工作站。大区所、地区所及工作站均属FDA的各级直属机构。区所的规模视工作量而定,全美的药品 65%以上在中大西洋区生产,故该区的力量较强,共有职工 525名,其中监督员 250名,约占FDA总部监督员的1/4,分析检验人员150名。 各州对药品的管理按地方药品管理法规进行,主要工作是:对药师进行考试和注册、对药品经营部门和药房进行监督检查,发放或换发许可证、吊销违法户的许可证、对所在地的药学院校进行评价、审查见习药房等。
http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif一局副局长胡凯红主持本次新闻发布会(刘健 摄)主持人胡凯红: 女士们、先生们,上午好,欢迎大家出席今天的新闻发布会。国务院《关于改革药品医疗器械审评审批制度的意见》已经正式印发。今天我们请来了国家食品药品监管总局副局长吴浈先生,请他向大家介绍药品医疗器械审评审批制度改革的有关情况,并回答大家的提问。2015-08-1809:54:47http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif国家食品药品监督管理总局副局长吴浈(刘健 摄)吴浈: 各位媒体朋友,大家上午好。今天国务院发布了《关于药品医疗器械审评审批制度的意见》,这个《意见》是8月13日印发的,各位朋友今天已经拿到正式的文件稿子。下面,我想给大家简单地介绍一下这次意见的起草背景和有关内容。2015-08-1809:57:07 吴浈: 第一,关于起草背景,为什么要起草这样一个改革意见。近年来,我们国家的医药产业发展比较快,药品医疗器械的质量和标准不断提高,公众用药需求得到了比较好的满足。但同时,药品和医疗器械审评过程当中存在的问题也越来越凸显,主要是现在药品审评积压比较严重,一些创新药品审评时间比较长,部分仿制药和国际先进水平还存在一定差距。出现这些问题,有很复杂的原因,既有历史的,也有机制的原因。大家都知道,我国曾经经历过药品短缺的年代,现代制药的起步也相对比较晚,标准相对偏低。这么多年来,国家采取的是将地方审评的药品集中到国家层面进行统一审批,药品标准由地方标准升为国家标准,提高GMP认证的水平,推进仿制药和原研药的一致性评价等等措施,药品质量明显提高,但是总体药品标准不够高,管理方式比较落后,审评审批体制不顺、机制不合理等等问题。企业低水平重复申报,部分注册申报的临床数据不真实,不完整、不规范,这些问题也越来越突出了,审批数量不够、待遇偏低,与审评的申报数量相比已经不匹配。2015-08-1809:57:48 吴浈: 党中央、国务院高度重视药品医疗器械审评审批制度改革。国务院关于改革药品医疗器械审评审批制度的意见,核心就是提高药品的质量,通过改革来促进医药行业产业的结构调整和转型升级,实现上市产品的有效性和安全性、质量可控性,能够达到国际先进水平,满足公众的用药需求,这就是这次起草这样一个改革方案的背景。2015-08-1810:01:18 吴浈: 改革意见主要内容,包括改革主要目标、主要任务、保障措施,一共三个方面的内容,总共21项改革措施。在改革目标上,我们重点围绕五个方面:一是提高药品审批质量;二是解决药品审评的积压;三是提高仿制药水平;四是要鼓励创新;五是要提高审评审批的透明度。大家手上有文件,我不一一展开作介绍了。 改革的主要任务12项,包括提高药品标准、推进仿制药一次性评价、加快创新药的审评审批,开展上市许可人持有制度的改革等等,总共12项任务。为了实现上述改革,国家食品药品监管总局将抓紧修订药品管理法实施条例,以及药品注册管理办法,面向社会招聘相应的药品审批技术人才,加强审评队伍建设,推进职业化检查员队伍建设。《意见》出台将对健全我国药品医疗器械审批审评体制和机制,提高药品审评审批质量和效率,提高上市药品的质量,促进医药产业创新和转型升级,将会起到积极作用。 我想把这些情况简单地给大家作个介绍,下面我很愿意回答大家的提问。2015-08-1810:02:15 主持人胡凯红: 谢谢吴局长的情况介绍,下面开始提问,提问之前先通报一下你所代表的新闻机构。2015-08-1810:03:25http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif中国国际广播电台记者提问(董金玲 摄) 中国国际广播电台记者: 我看到材料上说,我国药品注册申报积压比较严重,有没有具体数据说近年来到底一共积压了多少?是什么原因造成积压严重的问题?怎么解决?材料上说目标是2016年底消化完成注册积压的存量,这样严重的积压存量,如果想在明年年底之前消化完成,肯定要加快审批速度,会不会影响到药品质量的问题?2015-08-1810:03:38 吴浈: 当前药品的积压问题,总共积压了多少,国家药品审评中心正在进行审评的一共是21000件,和现在具体审评能力来讲,说实话任务量还是比较大,我们的能力和现实的审评量有比较大的差距。大家很关心,为什么有2100件?历史的背景和有关原因给大家做一个解读。药品审评积压,这是很复杂的过程,既有历史的原因,也有现实的问题,既有体制性的因素,也有机制性的情况,这些情况是交错在一起的。2015-08-1810:04:29 吴浈: 首先,药品注册审评积压是历史造成的。大家知道,药品审评在2000年以前都是由各省里承担,所以审评量都在各省分散。2000年以后,审评方式进行了改变,全部集中到中央来,因为药品审批事项中央事权,全国应该统一,不能分散,这是一个很大的改革,也是一个很好的进步。因此,把分散在各省的审评权力全部上收到中央来,审评量自然就增加了,但是相应的人员力量没有跟上,所以从上收以后,药品积压问题始终存在。 历史上出现过几次审评积压的高峰,比如说2005年,我们曾经手上积压过17000件,那时候审评人员只有100来人,采取了一些行政措施,削了峰。但是到了2007、2008年第二次高峰又来了,那时候积压了27000件。所以,2008年采取集中审评,解决了审评的积压问题,但是你看,从2011年以后,特别是近两年,又出现新的积压,现在到手上是21000件,这就叫三次高峰,三次波浪。2015-08-1810:05:06 吴浈: 我们尽管采取了一些有效措施来解决削峰问题,但是没有从根本上解决问题。所以,积压问题本身就是历史形成的,这里面有很多深层次的原因。比如现在的企业发展很快,但是产业基础又比较薄弱,低水平重复现象比较严重。我们国家应该说还是仿制药为主的国家,现在我们手上在审的21000个品种,90%是化药仿制药,化学药品里绝大部分,80%以上是仿制药。现在仿制药里面,水平不高,标准定得不高,现行法律规定,仿制药是仿现有国家标准,使得大家认为是仿标准,门槛就低了,门槛一低,申报量就大,申报量大的过程当中,重复率特别严重。2015-08-1810:07:06 吴浈: 我们有个数据,有八个品种,有100多家企业在申报,有23个品种,有50到99家申报。89个品种重复申报企业有20-49家。就这样三个数字,
监查员工作的重要意义和职责SFDA药品认证管理中心 曹 彩 一、SFDA药品认证中心管理职能:1、在SFDA统一部署下,参与制定、修订6个规章及其相应的管理办法。《药品非临床研究质量管理规范》(GLP)、《药品临床试验管理规范》(GCP)、《药品生产质量管理规范》(GMP)、《中药材生产质量管理规范》(GAP)、《药品经营质量管理规范》(GSP)、《医疗机构质量管理规范》(GUP)。其中GAP于2003年开始启动。2、SFDA药品认证管理中心职能: 受SFDA委托,组织对申请认证的药品研究机构、生产企业和医疗机构实施现场检查认证工作;3、承办对药品检查员的培训、考核和聘任及对省级药品监督管理部门的药品认证管理人员培训的具体工作;组织与6个规章相关的单位、企业的管理人员和技术人员的培训;4、受SFDA的委托,负责《药品认证公告》发布的具体工作;5、根据SFDA的安排,开展药品认证的国内、国际学术交流活动,承办国际间药品认证互认的具体工作;6、承办国家药品监督管理局交办地其他事项。
食品药品监督管理局局长陈锡江称目前正编写项目建议书 今日上线接受“问政”的镇街和单位:东莞市委政研室、石碣镇、东莞广播电视台、东莞报业传媒集团 东莞现有涉餐饮、药械、保健食品、化妆品单位有7万多家,让上千万人口吃上放心饭,用上安全药是一项艰巨任务。 昨日,在迎接党代会媒体集中采访会上,东莞市食品药品监督管理局局长陈锡江提到,抽检是监管的一个重要手段,东莞现在正在编写建设食品药品检测中心的项目建议书,拟投入1.5亿元。 而且,今年市政府将餐饮服务食品安全示范街创建工作列入了“十件实事”,这一工作目前已顺利完成。 【网友问政】 食品药品检测中心 启用未有时间表 市民潘先生:食品药品检测中心具体何时能启用? 陈锡江:食品药品检测中心规划占地25亩,投资1.5亿元,市政府已经基本同意,目前还在编制项目建议书,还没有到具体的实施阶段,所以还不能确定何时启用。 关键词 检测中心 规划面积25亩投资1.5亿元 技术监督一直是食品、药品监管的有力手段。陈锡江说,东莞不断拓宽检测领域,新增食品、保健食品、化妆品检验检测项目170项。在2006年与省医械所合作共建了生物性能实验室,合作范围逐年扩大。 但是,东莞技术监督目前却面临着场地问题。“东莞药品检验所在建立之初是全省最好的,但是,现在随着监管检测的新发展,面积已远远不够。”陈锡江说,建立东莞食品药品检测中心有着迫切的需求,“现在市政府已同意我们开始编写该项目建议书,其中规划面积为25亩,投资为1.5亿元。” 根据统计,东莞现有涉餐饮、药械、保健食品、化妆品单位7万多家,这也是保证上千万人口饮食用药安全的关键场所。 关键词 中毒事故 今年发生食物中毒事故仅7起 今年年初,东莞将餐饮服务食品安全示范街创建工作列入了“十件实事”,在全国率先开展创建工作。根据要求,要在今年年底全市至少有5条市级示范街创建完成并通过考核验收,力争不少于1条示范街达到省级标准并通过省的考核认定。 陈锡江说,目前,东莞已有15条创建街道全部通过示范创建考评,其中,2条街获得省级餐饮服务食品安全示范街称号。“可以说,‘十件实事’工作已经顺利完成,甚至超额完成了。” 而且,东莞示范街创建工作得到了国家食品药品监管局的充分肯定,本月28~30日,国家食品药品监督管理局、商务部将在东莞召开“全国餐饮服务食品安全示范工程建设现场会”,总结推广东莞创建做法、经验。 截至目前,东莞发生食物中毒事故7起,比去年同期有明显下降,以细菌性食物中毒为主,主要发生场所为集体食堂。
[b][font=黑体]一、项目基本情况[/font][/b][font=仿宋]项目编号:[/font][font=仿宋]HB2023113610050028[/font][font=仿宋]项目名称:[/font][font=仿宋]邯郸市疾病预防控制中心2023年结核病药品、试剂采购项目[/font][font=仿宋]预算金额:[/font][font=仿宋]1220000[/font][font=仿宋]最高限价(如有):[/font][font=仿宋]1220000[/font][font=仿宋]采购需求:[/font][font=仿宋]本项目共分为2个包,1包最高限价为1095000元;2包最高限价为125000元。[/font][font=仿宋]合同履行期限:[/font][font=仿宋]按需供货[/font][font=仿宋][font=仿宋]本项目[/font]不接受联合体投标。[/font][b][font=黑体]二、申请人的资格要求:[/font][/b][font=仿宋]1.满足《中华人民共和国政府采购法》第二十二条规定;[/font][font=仿宋]2.落实政府采购政策需满足的资格要求:本项目2包专门面向中小企业采购[/font][font=仿宋]3.本项目的特定资格要求:1包投标人如为生产厂家的,须具有药品生产许可证及所投产品对应的药品注册证;投标人如为代理商的,须具有药品经营许可证及所投产品对应的药品注册证复印件。2包投标人若为生产厂家,需具有《医疗器械生产许可证》、《医疗器械经营备案凭证》,若为代理商,需具有《医疗器械经营许可证》、《医疗器械经营备案凭证》,其产品应具有相对应的产品注册证(如属医疗器械的)。本项目不接受任何联合体投标。[/font][b][font=黑体]三、获取招标文件[/font][/b][font=仿宋]时间:[/font][font=仿宋]2023年12月01日至2023年12月07日,每天上午9:00至12:00,下午12:00至17:30[/font][font=仿宋](北京时间,法定节假日除外)[/font][font=仿宋]地点:[/font][font=仿宋]登录“邯郸市公共资源交易中心”网站自行免费下载招标文件[/font][font=仿宋]方式:[/font][font=仿宋]其它[/font][font=仿宋]售价:[/font][font=仿宋]0[/font][b][font=黑体]四、提交投标文件截止时间、开标时间和地点[/font][/b][font=仿宋]2023年12月21日14点00分[/font][font=仿宋](北京时间)[/font][font=仿宋]地点:邯郸市公共资源交易中心[/font][b][font=黑体]五、公告期限[/font][/b][font=仿宋]自本公告发布之日起[/font][font=仿宋]5个工作日。[/font][b][font=黑体]六、其他补充事宜[/font][/b][font=仿宋]提示:本项目按“双盲”评审要求招标,商务标(“明标”)和技术标(“暗标”)需分开制作,请投标人在“邯郸市公共资源交易中心”网站下载或更新最新版“投标文件制作工具”。[/font][b][font=黑体]七、对本次招标提出询问,请按以下方式联系。[/font][font=仿宋]1.采购人信息[/font][/b][font=仿宋]名[/font][font=仿宋] [/font][font=仿宋]称:[/font][font=仿宋]邯郸市疾病预防控制中心[/font][font=仿宋]地[/font][font=仿宋] [/font][font=仿宋]址:[/font][font=仿宋]邯郸市丛台区北仓路581号[/font][font=仿宋]联系方式:[/font][font=仿宋]0310-8168089[/font][b][font=仿宋]2.采购代理机构信息(如有)[/font][/b][font=仿宋]名[/font][font=仿宋] [/font][font=仿宋]称:[/font][font=仿宋]河北日进工程项目管理有限公司[/font][font=仿宋][font=仿宋]地[/font] [font=仿宋]址:[/font][/font][font=仿宋]邯郸市丛台区东柳北大街267号[/font][font=仿宋]联系方式:[/font][font=仿宋]0310-3102410[/font][b][font=仿宋]3.项目[/font][font=仿宋]联系方式[/font][/b][font=仿宋]项目联系人:[/font][font=仿宋]何运江[/font][font=仿宋][font=仿宋]电[/font] [font=仿宋]话:[/font][/font][font=仿宋]0310-3102410[/font]
药品的化学, 制造和控制(CMC):法规,质量,科学之要求及其策略研讨会举办城市: 上海举办地点: 上海美丽园龙都大酒店举办时间: 2010-6-28 ~ 2010-6-29主办单位: 美国药学科学家学会,和中国药学会药剂学专业委员会联系人: 倪正杰电话: 021-51691228-801传真: 021-63018596Email: zhliaust@online.sh.cn[font=宋体]由美国药学科学家学会(AAPS)和中国药学会(CPA)药剂学专业委员会联合主办的药品的化学、制造和控制(CMC)系列研讨会将于2010年6月28-29日在上海美丽园龙都大酒店召开,上海市食品药品监督管理局为本次研讨会的支持单位。研发出优质安全有效的药品并顺利注册申报是一个复杂的多元过程。药品的化学、制造和控制, 即CMC, 是产品成功开发并注册上市的最关键要素之一。这需要 (1)对产品各种研发活动中所涉及到的许多高度专业化学科的知识及问题有足够的理解 (2) 对各种研发实验及结果进行有效地链接 (3) 对全球区域性法规及其变化和发展趋势有良好理解 (4) 研发, 法规事务和质量部门间的紧密合作以制订法规申报策略和对研发数据的申报适用性进行评价。本次研讨会的演讲专家来自美国、中国药品监管部门以及国际著名制药企业,针对这些议题,来自法规、注册,质量和产品研发方面的专家们将在会上与大家分享他们的知识, 经验, 观察,认识和前瞻性看法,并提升企业拓展国际市场的能力。在会上,您可与制药行业的专家以及监管部门的官员共同探讨如何提升贵公司的研发和创新能力。本次研讨会将围绕以下几个主题展开充分的讨论:▲政府监管的法规框架、要求及期望(FDA美国食品药品监督管理局,ICH人用药物注册国际协调会议,CFR美国联邦法规、指南,质量,科学和GMP,质量源于设计/ICH Q8药物开发, Q9风险管理, Q10质量体系等几个指南,问答式评审,批准后变更,电子申报等)▲临床研究申请/新药申报/仿制药申报/通用技术文件/药物主控文件等文件中的CMC部分的结构和内容▲针对临床研究申请,新药申报和仿制药申报制订CMC对策和文件创建质量,GMP及合规性(厂房设施,质量体系,检查,供应链等)▲科学,质量和CMC策略和文件创建方面的工业界的实践,认识和看法▲全球化协作、整合和一体化的机遇[font=宋体]会议注册费(含资料费、28日中餐、晚宴、29日中餐及茶歇)企业工作人员: 人民币1600元(5月15日前缴费,可享8折优惠)政府工作人员: 人民币800元学生: 人民币800元(须出示有效证件,否则按正式代表收取)[font=宋体]附录一: 大会组委会荣誉主席郑梁元教授, 中国药学会药剂学专业委员会荣誉主席衣承东博士 上海市食品药品监督管理局副局长成员邱怡虹,高级研究员兼CMC业务副总监, 雅培制药, 会议兼主席张华, 上海市食品药品监督管理局认证审评中心首席认证员, 会议兼主席余煊强博士,美国FDA药品评价中心仿制药评审部科学和化学副部长, 会议兼主席陈琦琬,辉瑞公司全球CMC执行总监,前美国FDA新药质量评审部副部长张强教授,中国药学会药剂学专业委员会主席高惠君,上海市食品药品安全研究中心附录二:委员会成员和演讲者邮箱地址o Hua Zhang张华: zhanghua@smda.gov.cn or hzhang66@hotmail.com +86‐21‐6386‐7850o Qiang Zhang张强: zqdodo@bjmu.edu.cno TBD (SFDA)待定:o Brenda Uratani: brenda.uratani@fda.hhs.govo Guohua Zhang张国华: gh_zhang@yahoo.como Lawrence Yu余煊强: lawrence.yu@fda.hhs.govo Chi‐wan Chen陈琦琬: chi‐wan.chen@pfizer.como Richard Poska: richard.poska@abbott.como Jim Li 李建明: landin37@yahoo.como John Z. Duan 段宗一John.Duan@fda.hhs.govo Chengyi Liang梁诚一: chengyi.liang@fda.hhs.govo Weiqin (Tony) Tong童卫勤: Tony.Tong@tevausa.como Steve Colgan: stephen.t.colgan@pfizer.como He (Jim) Huang黄河: jhuang@actavis.como Ganapathy Mohan: ganapathy_mohan@merck.como Zi‐Qiang Gu顾自强: ziqiang.gu@fda.hhs.govo Sharon Pichon (AAPS): PichonS@aaps.org (+1‐703‐248‐4780)o Huijun Gao高惠君 (SHFDA): gaohj@smda.gov.cn or gaohj_1@126.com (021‐64511851:M:13816240286)o Yihong Qiu邱怡虹: qiu.yihong@abbott.com (+1‐847‐938‐5220 M : 847‐508‐9847)[/font][/font][/font]
中药临床试验申报资料中存在的问题SFDA药品审评中心审评一部许青峰一、对药品注册临床要求的理解: 总体要求、分类要求、受试例数二、试验常见问题试验设计、安全性数据总结分析三、对药品注册临床要求的理解(一)总体要求1、药物临床研究必须经国家药品监督管理局批准后实施,必须执行《药物临床试验质量管理规范》(GCP)2、药物的临床研究包括临床试验和生物等效性试验。3、申请新药注册、应当进行临床试验和生物等效性试验,申请已有国家标准的药品注册(仿制药),一般不进行临床研究,需用工艺和标准控制药品质量的中成药,应当进行临床试验。4、临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。申请新药注册应当进行Ⅰ、Ⅱ、Ⅲ期临床试验。有些情况下可仅进行Ⅱ期和Ⅲ期。或者Ⅲ期临床试验。5、药物临床研究的受试例数应当根据临床研究的目的,符合相关统计学的要求和本办法所规定的最低临床研究病例数要求。
药品质量受权人培训(国家食品药品监督管理局培训中心编)有些价值吧?由于附件PDF超过上传的50MB限度,所以只能分卷压缩,只有全部下载下来才能打开!!!
药品注册,是国家食品药品监督管理局依照《药品管理法》的规定,根据药品注册申请人的申请,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。在药品研制、生产、流通、使用的全过程监管中,药品注册管理是从源头上对药品安全性和有效性实施监管的重要手段,其根本目的是通过科学评价,保证上市药品安全有效,保障和促进公众健康。 1 2010年药品注册管理的重要举措 2010年,药品注册管理工作继续践行科学监管理念,紧紧围绕“质量和效率”这个中心,以风险效益评估和风险管理为核心,坚持“规范审批、公开透明、鼓励创新”的原则,全面推进体制机制改革和法制建设,强化学药品物研究全程监管,进一步推进审评审批公开公平,提高审评审批效率,较好地履行了《药品管理法》赋予的职责。 1.1 完善药品注册管理法规体系 一是发布了《药物临床试验伦理审查工作指导原则》,规范和指导伦理委员会的药物临床试验伦理审查工作,加强药物临床试验的质量管理和对受试者的保护,提高药物临床试验伦理审查工作质量。继续开展《药用原辅材料登记备案管理规定》、《药品标准管理办法》、《药物临床试验生物样本分析实验室管理规定》、《药物Ⅰ期临床试验管理指导原则》、《药物临床试验中严重不良事件报告与监测管理规定》及天然药物注册技术要求等法规和规范性文件的研究起草工作。
[size=4] 四月的洛阳牡丹绽放,繁花似锦,珍奇斗艳。由国家科技部批准以洛阳惠中集团为依托的国家兽用药品工程技术研究中心落户普莱柯科技园,该中心的奠基仪式于4月13日上午8:30在普莱柯科技园举行。科技部党组成员、纪检组郭向远组长,科技部发展计划司王晓方司长,农业部兽医局林典生处长,中国畜牧兽医学会副理事长兼秘书长阎汉平研究员,中国动物保健品协会马保环秘书长,河南省科技厅赵琛厅长,河南省畜牧局朱立良副局长,洛阳市郭洪昌市长以及洛阳高新区王忠道主任等领导出席奠基仪式并发表重要讲话。应邀莅临此次奠基仪式的嘉宾还有来自其他省市畜牧兽医局的领导、河南农大、中牧集团、新闻媒体和普莱柯战略联盟商等。[/size][size=4] 在奠基仪式上,惠中集团董事长张许科研究员详细地汇报了集团在申报此项工程中所做的工作。洛阳惠中集团包括惠中兽药、普莱柯和郑州新正好等GMP企业,集团是首批认定的新标准高新技术企业和河南省政府重点支持的50家高成长型高新技术企业,其技术创新与产品研发处于业界领先地位,并在品牌影响力、生产规模、市场开拓、经济效益等综合实力方面居我国兽用药品行业前列。集团业已高效运行两大科技创新平台,河南省兽药工程技术研究中心和以该中心为主导的、与中国农业大学、美国SBC公司等国内外17家院所建立的紧密的产学研战略联盟,相继攻克了药物缓释、分子包被、细胞克隆与悬浮培养等一批产业支撑核心技术,国内率先研制成功头孢噻呋、泰拉霉素,禽流感H9三联灭活疫苗、高效猪瘟疫苗、猪圆环病毒2型灭活疫苗等新型兽用药品,取得了不小的业绩,集团原创新兽药占2003年以来全国原创新兽药批准总数的10%以上。基于惠中集团的技术创新与产品研发实力,科技部于2009年10月正式批准了依托该集团公司组建国家兽用药品工程技术研究中心的申请,该中心成为我国兽用药品领域唯一获准的国家级研究中心。[/size][size=4] 科技部王晓方司长肯定了该中心的建设将对我国发展规模养殖业和推动兽用药品行业进步的重要作用和意义,对促进养殖健康、改善食品安全将大有作为,并作为科技推动生产的典范予以推广;河南省科技厅领导也肯定了集团以往取得的业绩并希望今后再接再厉,再创新佳绩;省市其他领导还表扬了集团作为民营企业能够在科技研发上大力投入,创立自主知识产权所做出的贡献。[/size][size=4] 按规划,国家兽用药品工程技术研究中心计划投资6300万元,建筑面积13422平方米,预计2011年底竣工。该中心建成后将针对兽用原料药、高新制剂、中兽药新产品、生物制品等方面开展关键技术研究、成果转化中的产业技术研究,同时具备承接政府、企业、院所委托的开发任务以及对外技术服务的能力,以切实提升我国兽用药品行业技术的整体创新能力和核心竞争力。[/size]
中新网8月8日电 今天上午,国家食品药品监督管理局举行例行新闻发布会。据新闻发言人颜江瑛介绍,国家药监局着手研究和实施“食品药品监管系统基础设施建设规划项目”。项目总投资88亿元,中央政府投资63亿元,地方政府投资25亿元。 项目目标是:争取用3到5年时间,通过“食品药品监督管理系统基础设施的项目”的实施,中央级基础设施达到与食品药品监管工作、经济社会发展相适应,技术手段在国际上处于比较先进的水平;地方的食品药品监管体系能够承担法定赋予的各项任务。“食品药品监管系统基础设施建设规划项目”主要包括以下六个内容: 1. 中国药品生物制品检定所迁建项目 中国药品生物制品检定所在现址上的占地面积近6.7万平方米,建筑面积5.82万平方米,其中实验室4.2万平方米。由于现有的场地不能满足业务发展的需要,而且药检所目前所在地又是北京市要求限期搬迁的地方。因此,结合《国家高级别生物安全实验室体系建设规划》和新扩展业务所需的实验室项目建设,一并考虑迁建药检所。中国药品生物制品检定所搬迁项目的主要建设内容有:药品检验楼、生物制品检验楼、生物安全实验楼、生物诊断试剂检验楼、动物中心楼、综合业务楼、信息楼、科技楼及毒种、菌种等附属用房等。迁建项目总建筑面积约9.2万平方米,占地200亩。目前已确定建设地点,正在进行环境评价等工作。 2. 国家口岸药检所改造项目 国家食品药品监督管理局指定中国药品生物制品检定所和16个地方药检所作为进口药物的口岸药检所。目前,16个口岸所不具备进口药品全项检验能力,特别是对生物制品和药包材的检验能力不够,动物实验室的能力也普遍不足。国家口岸药检所改造项目的建设内容为:16个口岸药检所的生物检测设施改造。实验室改造面积共计1.8万平方米,配备改造实验室所需的实验室设备,并依据《全国药品检验机构基本仪器配置标准》,增补检测设备。目前,项目可行性研究报告正在审批中。 3.国家医疗器械检测中心改造项目 国家先后设立了10个医疗器械质量检验中心,分管不同品种的医疗器械检验工作。按各中心的专业分工,项目重点对全国10个医疗器械检测中心的专业检测室进行改造、配备相应仪器设备。根据医疗检测产品的专业需要和归口管理的要求,结合我国医疗器械生产的特性、生产厂家的地域和分布密度,建议适当的电磁兼容安全检测室,配备相应的检测设备,建筑面积9.8万平方米。目前,可行性研究报告即将编制完成。 4.国家药品不良反应检测体系建设项目 项目建设内容包括一个国家药品不良反应检测中心和31个分中心,建立药品不良反应(包括进口药物)、医疗器械(包括进口医疗器械)不良事件及药物滥用监测网络信息系统,网络信息系统联接国家中心和31个分中心。国家中心是国家局的直属事业单位,与药品评价中心属两块班子一班人马,不再建设业务用房;31个分中心分别设在各省,解决工作必需的业务用房,分中心工作由省级食品药品监管部门负责。目前,可行性研究报告正在审批中。 5.西部地区食品药品监管行政执法机构基础设施建设西部地区药检所改造项目 项目建设内容主要包括:西部地区省、地县三组行政执法机构办公业务用房建设,建筑面积共46万平方米;按《全国药品监督管理机构执法基本装备标准》对西部地区行政机构执法装备进行补充配备;对西部地区省、地两级药检所的改造,改造实验室面积10万平方米,并按《全国药品检验机构基本仪器配置标准》,增补相应的检测仪器和专用设备。目前,办公业务用房建设和实施室改造已全面实施。仪器设备配备实施方案正在审批。 6.中部地区食品药品监管行政执法机构基础设施建设及中部地区药检所改造项目 项目建设内容主要包括:中部地区省、地、县三级行政执法机构办公业务用房建设,建筑面积共48万平方米;按《全国药品监督管理机构执法基本装备标准》对中部地区行政执法机构执法装备进行补充配备;对中部地区省、地两级药检所的改造,改造实验室面积约13万平方米,并按《全国药品检验机构基本仪器配置标准》,增补相应的检测仪器和专用设备。目前,办公业务用房建设和实施室改造已全面实施。仪器设备配备实施方案正在审批。 东部地区食品药品监管行政执法机构基础设施建设和药检所改造原则上由地方政府自行解决。 另外,中央和地方政府还将根据《国家食品药品安全“十一五”规划》的食品和药品安全建设项目,在“十一五”期间相应的投入也将分别到位。(据中国网文字直播整理)
原创作品评分是一件很严肃的事情,也是很容易引起争执的事件。本次原创作品评优分为两个部分,月度评优、年度评优。月度评优分为两个部分:票选、评委团评分。根据评委团评分每个评委的得分,计算出评委团评分;再加上投票分,最后得出作品最终得分。根据赛区文章数量,按一定获奖比例确定获奖作品数量;依据获奖数量,确定一、二、三等奖各级奖项的获奖名额。根据赛区内作品最终得分排名,确定各个奖项对应的作品。对大家争议比较大的评委评分,采取各种相应措施,尽可能保持评分的客观性。首先确立评分标准。评分标准主要分为两个部分:内容的完整性和创新性。如果按照评分标准,没有创新的文章,直接就扣去了30分,剩余70分能获大奖吗?但是,有创新就一定获奖吗?逻辑混乱、话不成句的作品,按评分标准,能获奖吗?从大家反映的意见,对此评分标准,并无很大异议。接下来是如何按标准进行评分。为了避免个人主观意见对作品评分的影响,引进评审团。每个赛区评委数量基本保证3个以上,多者5人。基本保证一篇作品的最终评分不会偏离大的区间。网友投票,这可以看出这篇作品的受欢迎程度。也许这作品,创新内容不足,内容完整性也有所欠缺,但内容喜闻乐见,在评审团得分的基础上,加上投票得分,也有助于此类作品名次的提高。从目前情况看,也许,最重要的不是标准是什么,最重要的是用同一个标准去评价作品。如何实现这一目标,也许才是诸位最关心的问题;也是我们需要大家多提意见,以便根据情况能够不断加以改进的地方。
[quote][b]项目概况[/b]吉林市生态环境监控中心购置试剂、实验药品及材料项目 招标项目的潜在投标人应在ggzy.jlcity.gov.cn获取招标文件,并于2022年11月17日 13点30分(北京时间)前递交投标文件。[/quote][font=inherit]一、项目基本情况[/font]项目编号:JSZFCG—J2022ZX088项目名称:吉林市生态环境监控中心购置试剂、实验药品及材料项目预算金额:96.0000000 万元(人民币)最高限价(如有):0.0000000 万元(人民币)采购需求:购置试剂、实验药品及材料(具体内容请下载招标文件及投标报价明细表)。合同履行期限:按招标文件规定时间履行。本项目( 不接受 )联合体投标。
昨天(27日),国家食品药品监督管理局副局长吴浈做客中国网。他表示,今年国家药监局将采取评审人员集体负责制以及审批责任追究制等办法完善药品审批制度,防止腐败。 “三制一化”防止腐败 由于郑筱萸案暴露出药品审批中的腐败,昨天吴浈表示,药品审批过程当中出现了一些腐败现象,应该要正视。虽然是少数人,但已让整个系统蒙羞。他说,今年国家药监局会加强药品审评审批的管理,在药品审评审批中实行“三制一化”。所谓“三制一化”,就是实行审评人员集体负责制,目的就是防止个别人滥用审评审批权力;实行审评审批责任追究制,强化对违法审批行为的责任追究;实行审评审批检验人员公示制,将药品审评审批置于社会监督之下。他说,目前国家药监局正在全力抓药品审批的信息平台建设,目的就是让审批能够公开透明。今后,国家药监局要逐步实行药品审批在网上进行受理。 驻厂监督员无权处罚企业 由于不久前媒体广泛报道了国家药监局将向药厂派驻驻厂监督员,吴浈说,今天第一批派驻监督员将在国家药监局培训。他强调,驻厂监督员不包揽企业的质量责任,企业永远是产品质量的第一责任人。同时,驻厂监督员无权直接处罚企业。 02年批号药品今年再注册 对于国家药监局日前提出的药品再注册,吴浈解释说,药品再注册是对药品批准证明文件有效期满后要求继续生产药品实施再审批过程。根据规定,药品批准文号的有效期都是5年。有效期满以后,都必须按照有关规定进行药品再注册。 目前再注册的范围是,2002年换发批准文号的品种以及2003年新批准上市的品种进行再注册,因为这些药有效期已满5年。并不是所有药品都要再注册。
[quote][b]项目概况[/b]吉林市生态环境监控中心购置试剂、实验药品及材料项目 招标项目的潜在投标人应在ggzy.jlcity.gov.cn获取招标文件,并于2023年02月15日 09点10分(北京时间)前递交投标文件。[/quote][font=inherit]一、项目基本情况[/font]项目编号:JSZFCG—J2022ZX088项目名称:吉林市生态环境监控中心购置试剂、实验药品及材料项目预算金额:96.0000000 万元(人民币)最高限价(如有):0.0000000 万元(人民币)采购需求:购置试剂、实验药品及材料(具体内容请下载招标文件及投标报价明细表)合同履行期限:按招标文件规定时间履行本项目( 不接受 )联合体投标。
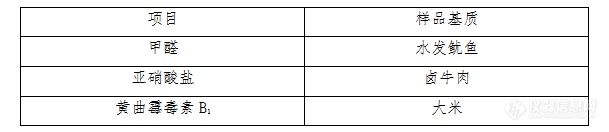
食品安全快速检测产品评价技术报告根据《福建省食品药品监督管理局办公室关于做好食品快速检测产品评价工作的通知》(闽食药监 办科函(2017)202号),结合我市今年快检工作实际,我中心于2018年01月对我市已投入使用的快检产品按照总局《食品快速检测方法评价技术规范》和省局《食品快速检测产品评价工作参考指南》的要求开展评价工作,现报告如下: 一、评价对象厦门斯坦道科学仪器股份有限公司 快检仪器:STD-9000斯坦道科学仪器 及相关产品的快检试剂盒。 二、评价依据 1、食品快速检测方法评价技术规范(食药监办科〔2017〕43号) 2、总局关于规范食品快速检测方法使用管理的意见(食药监科〔2017〕49号) 3、总局发布的食品快速检测方法的公告(2017第58号KJ201704、2017第92号KJ201708) 三、评价项目 1、甲醛 2、亚硝酸盐 3、黄曲霉毒素B1aa. 黄曲霉毒素B1项目快检仪器厂商只提供试剂盒法,同一快检设备的试剂盒均相同,仅作一次评价。 四、评价指标 1、形式检查 包括产品的包装、标签、标识、使用说明、质量合格证书等资料。 2、方法评价指标 包括灵敏度、特异性、假阴性率、假阳性率和与参比方法的一致性等。 五、评价方法 1、盲样制备 按照《福建省食品药品监督管理局办公室关于做好食品快速检测产品评价工作的通知》(闽食药监 办科函(2017)202号)附件《食品快速检测产品评价工作参考指南》,根据供应商产品标称的适用范围,选取一类典型样品,每家共制备阴性样品20个,阳性样品20个,其中10个阴性样品和10阳性样品用于快检测定,阳性样品目标物添加浓度水平为产品检测水平,按照供应商提供的试剂、方法和仪器进行,另外10个阴性样品和10个阳性样品按照相应的国标方法进行。所有的阴性样品经参比方法确认不含目标物,阳性样品经参比方法测定结果与实际样品一致。具体选取样品基质如下:[img=,609,132]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051506386713_7673_2166779_3.png!w609x132.jpg[/img]2、结果判定 按照总局食品快速检测方法评价技术规范(食药监办科〔2017〕43号)的要求,对方法的灵敏度、特异性、假阴性率、假阴性率进行评价,结果应符合总局公布的快检方法中的性能指标要求,快检方法与参比方法的经卡方检验,阳性确证比率在95%的置信区间内没有显著性差异,详见附件1.六、测定结果1、甲醛(批号STD2017122801):,结果见表1.[img=,690,612]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051507592673_1690_2166779_3.png!w690x612.jpg[/img] 2、亚硝酸盐(批号STD2018011201):结果见表2. 表2 卤牛肉中亚硝酸盐测定结果[img=,638,590]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051509352147_3733_2166779_3.png!w638x590.jpg[/img]3、试剂盒黄曲霉毒素B[sub]1[/sub](批号STD2017122102):结果见表3:表3 大米中黄曲霉毒素B[sub]1[/sub]测定结果[img=,645,591]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051511543247_412_2166779_3.png!w645x591.jpg[/img][img=,690,408]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051512019913_5283_2166779_3.png!w690x408.jpg[/img][img=,690,478]http://ng1.17img.cn/bbsfiles/images/2018/08/201808051512088957_897_2166779_3.png!w690x478.jpg[/img]
根据新华社北京4月24日电,第十二届全国人民代表大会常务委员会第十四次会议决定从即日起对《中华人民共和国药品管理法》作了相应修改,重新公布。这次修改涉及生产经营活动、资质资格类审批的取消,取消有关工商登记前置审批权,以及放开药品定价权等事项。这将意味着企业将真正成为市场的主体,将要有更强的市场应变力和竞争力,才能立足于市场。附《第十二届全国人民代表大会常务委员会第十四次会议决定》和药品管理法删除部分内容第十二届全国人民代表大会常务委员会第十四次会议决定对《中华人民共和国药品管理法》作如下修改: 一、删去第七条第一款中的“凭《药品生产许可证》到工商行政管理部门办理登记注册”。 二、删去第十四条第一款中的“凭《药品经营许可证》到工商行政管理部门办理登记注册”。 三、删去第五十五条。 四、将第八十九条改为第八十八条,并删去其中的“第五十七条”。 五、删去第一百条。 本决定自公布之日起施行。 《中华人民共和国药品管理法》根据本决定作相应修改,重新公布。药品管理法删除的部分内容 第五十五条 依法实行政府定价、政府指导价的药品,政府价格主管部门应当依照《中华人民共和国价格法》规定的定价原则,依据社会平均成本、市场供求状况和社会承受能力合理制定和调整价格,做到质价相符,消除虚高价格,保护用药者的正当利益。药品的生产企业、经营企业和医疗机构必须执行政府定价、政府指导价,不得以任何形式擅自提高价格。 药品生产企业应当依法向政府价格主管部门如实提供药品的生产经营成本,不得拒报、虚报、瞒报。 第五十七条 药品的生产企业、经营企业、医疗机构应当依法向政府价格主管部门提供其药品的实际购销价格和购销数量等资料。 第一百条 依照本法被吊销《药品生产许可证》、《药品经营许可证》的,由药品监督管理部门通知工商行政管理部门办理变更或者注销登记。
[color=#cc0000][b]什么是重新评审?什么是重新采购?[/b][/color][color=#cc0000][b] 重新评审:单纯指针对某个政府采购项目,由于出现了非正常流程的因素,导致原评审结果无效,而必须对原来的文件再一次邀请专家来就某一项重新做出评定,以对采购结果进行重新评价,从而完成本次采购流程的评审活动。[/b][/color][color=#cc0000][b][/b][/color][color=#cc0000][b] 重新采购:即本次项目采购结果无效,相关流程均须重新走一遍,如需要重新制作采购文件,需要重新发布招标公告、竞争性谈判邀请、竞争性磋商邀请、询价邀请、单一来源邀请等,其范畴要更大一些,包括了重新评审这一环节。[/b][/color]
食品药品监管总局组织制定了化学药品注册分类改革工作方案(以下简称方案),已于2016年3月4日正式发布实施。现就有关问题解读如下:[b]一、方案的出台背景。[/b] 2015年8月9日,国务院发布《关于改革药品医疗器械审评审批制度的意见》,明确调整药品注册分类。2015年11月4日,第十二届全国人民代表大会常务委员会第十七次会议审议通过《关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定》,同意国务院组织开展药品注册分类改革。为落实上述政策要求,满足实际工作需要,食品药品监管总局制定发布了本方案。[b]二、方案的适用范围。[/b] 本方案仅针对化学药品注册分类进行了调整,适用于方案发布实施后新受理的化学药品注册申请(包括临床、生产、进口注册申请)。[b]三、新注册分类中,关于新药的含义是什么?[/b] 新药指中国境内外均未上市的药品,分为创新药和改良型新药。 新注册分类1为创新药,强调含有新的结构明确的、具有药理作用的化合物;新注册分类2为改良型新药,在已知活性成份基础上进行优化,强调具有明显的临床优势。[b]四、创新药具体包括什么?[/b] 创新药指含有新的结构明确、具有药理作用的化合物,且具有临床价值的药品,不包括改良型新药中2.1类的药品。[b]五、新注册分类中,关于仿制药的含义是什么?[/b] 仿制药是指仿制已上市原研药品的药品,分为两类,一是仿制境外已上市境内未上市原研药品,二是仿制境内已上市原研药品。仿制药要求与原研药品质量和疗效一致。 如果已上市药品的原研药品无法追溯或者原研药品已经撤市的,建议不再申请仿制;如坚持提出仿制药申请,原则上不能以仿制药的技术要求予以批准,应按照新药的要求开展相关研究。[b]六、新注册分类中,关于第5类药品的含义是什么?[/b] 第5类药品是指境外上市的药品申请在中国境内上市,分为原研药品和非原研药品两类。对应原化学药品注册分类中的进口药品类别。[b]七、含有未知活性成份的新复方制剂按什么申报?[/b] 含有新的结构明确的、具有药理作用的化合物的新复方制剂,应按照新注册分类1进行申报。[b]八、仿制药是否要求处方工艺、规格、用法用量等均与原研保持一致?[/b] 仿制药要求与原研药品具有相同的活性成分、剂型、规格、适应症、给药途径和用法用量,不强调处方工艺与原研药品一致,但强调仿制药品必须与原研药品质量和疗效一致。[b]九、新注册分类下的技术标准有何变化?[/b] 对按新注册分类申报的化学药品注册申请实行新的审评技术标准。其中,对于创新药,一是强调“创新性”,即应当具备“全球新”的物质结构,二是强调药物具有临床价值;对于改良型新药,强调“优效性”,即相较于被改良的药品,具备明显的临床优势;对于仿制药,强调“一致性”,被仿制药品为原研药品,且质量与疗效应当于原研药品一致。[b]十、新注册分类的监测期设置是如何考虑的?[/b] 《中华人民共和国药品管理法实施条例》第三十四条规定,国务院药品监督管理部门根据保护公众健康的要求,可以对药品生产企业生产的新药品种设立不超过5年的监测期。新注册分类中的第1类、第2类属于新药范畴,因此设置监测期。同时,参照《药品注册管理办法》中原化学药品注册分类的监测期期限设置,对于新注册分类中与原注册分类相类似或者相近的情形分别设置了相应的监测期限。[b]十一、已经受理的原注册申请在新注册分类实施后如何过渡?[/b] 方案发布实施后新申报的化学药品注册申请(包括临床、生产、进口申请),其注册分类按照新注册分类有关要求执行。 方案发布实施前已受理尚未完成审评审批的化学药品第3类及6类报生产注册申请,申请人可以选择继续按照原规定进行审评审批,符合要求的,在批准时,注册分类项注明相应的原注册分类;申请人也可以申请按照新注册分类进行后续申报、审评审批。 对于申请按照新注册分类进行审评审批的注册申请,由食品药品监管总局药审中心优先按照新注册分类的要求加快审评审批;经审查不符合要求的,不再要求补充资料,直接不予批准。[b]十二、方案中“补交相关费用”具体如何执行?[/b] 方案“二、相关注册要求”中第(四)部分提及的“补交相关费用”是指,2015年5月27日前已经受理的化学药品注册申请,申请按照新注册分类进行审评审批的,应当按照《关于发布药品、医疗器械产品注册收费标准的公告》(2015年第53号)公布的《药品、医疗器械产品注册收费标准》补交相关注册收费;2015年5月27日以后受理的、已按照上述标准交费的化学药品注册申请,申请按照新注册分类进行审评审批的,无须补交费用。[b]十三、新注册分类实施后,后续有何细化工作要求?[/b] 新注册分类实施后,食品药品监管总局将组织相关部门尽快提出《新注册分类申报资料项目及要求》、《新注册分类受理审查指南》、《新注册分类核查检查要点》、《新注册分类技术审评指导原则》等在内的一系列相关细化工作要求,以便指导申请人合理研发申报相关产品,进一步提高药品质量。
[table=100%][tr][td]项目概况:[/td][/tr][tr][td] 汶上县检验检测中心药品试剂、耗材采购采购项目的潜在供应商应在中国山东政府采购网(www.ccgp-shandong.gov.cn)或济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)获取采购文件,并于2023-08-15 09:30:00(北京时间)前提交响应文件。[/td][/tr][/table][font=&][size=16px][color=#383838]一、项目基本情况:[/color][/size][/font][font=&][size=16px][color=#383838] 项目编号:SDGP370830000202302000117 项目名称:汶上县检验检测中心药品试剂、耗材采购 采购方式:竞争性磋商 预算金额:46.8212万元 最高限价:46.8212万元 采购需求:[/color][/size][/font][table=100%][tr][td]标的[/td][td]标的名称[/td][td]数量[/td][td]简要技术需求或服务要求[/td][td]本包预算金额(单位:万元)[/td][/tr][tr][td]A[/td][td]标准物质和试剂 [/td][td]1 [/td][td]1.满足《中华人民共和国政府采购法》第二十二条规定;(1)具有独立承担民事责任的能力并满足磋商文件要求的供应商;(2)须遵守《中华人民共和国政府采购法》及相关法律、法规和规章;(3)单位负责人为同一人或者存在控股、管理关系的不同单位,不得同时参加同一项目或同一包组的投标;生产厂家的母公司、全资子公司、控股公司,只能有一家参加同一招标编号的投标;为本项目提供整体设计、规范编制或者项目管理、监理、检测等服务的投标供应商,不得再参加该本项目的其他采购活动;(4)具备有效的营业执照,并在人员、设备、资金等方面具有相应的能力;(5)供应商须具有良好的商业信誉和健全的财务会计制度、有依法缴纳税收和社会保障资金的良好记录;(6)供应商信用状况良好,未被列入经营异常名录或者严重违法企业名单,不得存在相关行贿犯罪记录;2.落实政府采购政策需满足的资格要求:根据《政府采购中小企业发展管理办法》第六条第三款“符合下列情形之一的,可不专门面向中小企业预留采购份额:(三)按照本办法规定预留采购份额无法确保充分供应、充分竞争,或者存在可能影响政府采购目标实现的情形;”之规定,本项目不专门面向中小企业预留采购份额;3.本项目的特定资格要求:无;4.本项目兼投兼中;5.本项目为资格后审。 [/td][td]46.821200 [/td][/tr][/table][font=&][size=16px][color=#383838] 合同履行期限:接到采购人通知后2日内供货完毕。 本项目不接受联合体投标。[/color][/size][/font][font=&][size=16px][color=#383838]二、申请人的资格要求:[/color][/size][/font][font=&][size=16px][color=#383838] 1、满足《中华人民共和国政府采购法》第二十二条规定; 2、落实政府采购政策需满足的资格要求:根据《政府采购中小企业发展管理办法》第六条第三款“符合下列情形之一的,可不专门面向中小企业预留采购份额:(三)按照本办法规定预留采购份额无法确保充分供应、充分竞争,或者存在可能影响政府采购目标实现的情形;”之规定,本项目不专门面向中小企业预留采购份额; 3、本项目的特定资格要求:无[/color][/size][/font][font=&][size=16px][color=#383838]三、获取采购文件:[/color][/size][/font][font=&][size=16px][color=#383838] 1.时间:2023年8月4日17时30分至2023年8月14日15时0分,每天上午00:00至11:59,下午12:00至23:59(北京时间,法定节假日除外 ) 2.地点:中国山东政府采购网(www.ccgp-shandong.gov.cn)或济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang) 3.方式:自主下载 4.售价:0[/color][/size][/font][font=&][size=16px][color=#383838]四、响应文件提交:[/color][/size][/font][font=&][size=16px][color=#383838] 1.截止时间:2023年8月15日9时30分(北京时间) 2.地 点:所有响应文件应于2023年8月15日9时30分(北京时间)之前上传到济宁市公共资源交易服务中心汶上分中心网,未在规定时间内上传响应文件电子文档或截止时间后上传的电子文档将不予公开唱标及评审。[/color][/size][/font][font=&][size=16px][color=#383838]五、开启:[/color][/size][/font][font=&][size=16px][color=#383838] 1.开启时间:2023年8月15日9时30分(北京时间) 2.开启地点:登录济宁市公共资源交易网不见面开标大厅进行解密[/color][/size][/font][font=&][size=16px][color=#383838]六、公告期限:[/color][/size][/font][font=&][size=16px][color=#383838] 自本公告发布之日起3个工作日。[/color][/size][/font][font=&][size=16px][color=#383838]七、其他补充事宜:[/color][/size][/font][font=&][size=16px][color=#383838] 其他补充事宜:1.项目参与:如潜在供应商予参与本项目,须同时在中国山东政府采购网(www.ccgp-shandong.gov.cn)、济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)登录、注册,响应本项目。未按要求响应本项目的,按无效报价处理。2.具体响应如下:(1)济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)—用户登录——政府采购,响应本项目并下载文件。【凡未在济宁市公共资源交易服务中心汶上分中心注册的供应商应先办理注册(济宁市公共资源交易服务中心汶上分中心网站-用户注册-政府采购)。所有项目全流程电子化并无任何纸质文件,所以注册后必须办理CA及电子签章,CA办理及系统操作咨询:0537-7817075、0537-6050123。已办理过注册及电子签章的供应商,登录系统,参与本项目。】(2)中国山东政府采购网(www.ccgp-shandong.gov.cn)-用户登录点击报名,报名后没有其他操作,无须在该网站上传电子版响应文件及报价。供应商未注册的先进行注册。(3)济宁市公共资源交易服务中心汶上分中心网与济宁市公共资源交易服务中心网在注册,登录、响应项目操作过程是通用的。各供应商在投标前须办理企业网上注册手续,具体程序详见济宁市公共资源交易服务中心《政府采购注册流程》。3.电子版文件上传1)本项目实行网上报价和上传响应文件电子文档所有响应文件应于2023年8月15日9时30分(北京时间)之前上传到济宁市公共资源交易服务中心汶上分中心网,未在规定时间内上传响应文件电子文档或截止时间后上传的电子文档将不予公开唱标及评审。2)具体操作:登录“济宁市公共资源交易服务中心汶上分中心网”用户登录政府政府采购,点击项目,上传文件。4.本项目公告将同时在济宁市公共资源交易服务中心汶上分中心网、中国山东政府采购网发布。本项目如有必要澄清和修改需要发布更正公告(变更公告)的,将同时在济宁市公共资源交易服务中心汶上分中心网、中国山东政府采购网及时发布,请各潜在供应商及时关注相关信息。更正公告(变更公告)一旦发布即视为通知所有潜在供应商。5.本项目为资格后审。6.本项目共分为2个包组。按包组先后顺序确定拟成交包组,开标、评标顺序按照项目包组顺序。包组开标顺序:A包、B包依次开标。包组评审顺序:A包、B包依次评审。请参与响应的企业认真核对所响应包组,避免上传文件出现失误。[/color][/size][/font][font=&][size=16px][color=#383838]八、对本次招标提出询问,请按以下方式联系:[/color][/size][/font][font=&][size=16px][color=#383838] 1、采购人信息 名 称:[/color][/size][/font][font=&][size=16px][color=#383838]汶上县检验检测中心 地 址:(汶上县检验检测中心) 联系方式:1668805347 2、采购代理机构 名 称:[/color][/size][/font][font=&][size=16px][color=#383838]山东政联建设项目管理有限公司 地 址:山东省济宁市高新区县(区)洸河百丰商贸中心号十九层 联系方式:15563772976 3、项目联系方式 项目联系人:孔芬 联系方式:15563772976[/color][/size][/font][table=100%][tr][td]项目概况:[/td][/tr][tr][td] 汶上县检验检测中心药品试剂、耗材采购采购项目的潜在供应商应在中国山东政府采购网(www.ccgp-shandong.gov.cn)或济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)获取采购文件,并于2023-08-15 09:30:00(北京时间)前提交响应文件。[/td][/tr][/table][font=&][size=16px][color=#383838]一、项目基本情况:[/color][/size][/font][font=&][size=16px][color=#383838] 项目编号:SDGP370830000202302000117 项目名称:汶上县检验检测中心药品试剂、耗材采购 采购方式:竞争性磋商 预算金额:46.6518万元 最高限价:46.6518万元 采购需求:[/color][/size][/font][table=100%][tr][td]标的[/td][td]标的名称[/td][td]数量[/td][td]简要技术需求或服务要求[/td][td]本包预算金额(单位:万元)[/td][/tr][tr][td]B[/td][td]耗材 [/td][td]1 [/td][td]1.满足《中华人民共和国政府采购法》第二十二条规定;(1)具有独立承担民事责任的能力并满足磋商文件要求的供应商;(2)须遵守《中华人民共和国政府采购法》及相关法律、法规和规章;(3)单位负责人为同一人或者存在控股、管理关系的不同单位,不得同时参加同一项目或同一包组的投标;生产厂家的母公司、全资子公司、控股公司,只能有一家参加同一招标编号的投标;为本项目提供整体设计、规范编制或者项目管理、监理、检测等服务的投标供应商,不得再参加该本项目的其他采购活动;(4)具备有效的营业执照,并在人员、设备、资金等方面具有相应的能力;(5)供应商须具有良好的商业信誉和健全的财务会计制度、有依法缴纳税收和社会保障资金的良好记录;(6)供应商信用状况良好,未被列入经营异常名录或者严重违法企业名单,不得存在相关行贿犯罪记录;2.落实政府采购政策需满足的资格要求:根据《政府采购中小企业发展管理办法》第六条第三款“符合下列情形之一的,可不专门面向中小企业预留采购份额:(三)按照本办法规定预留采购份额无法确保充分供应、充分竞争,或者存在可能影响政府采购目标实现的情形;”之规定,本项目不专门面向中小企业预留采购份额;3.本项目的特定资格要求:无;4.本项目兼投兼中;5.本项目为资格后审。 [/td][td]46.651800 [/td][/tr][/table][font=&][size=16px][color=#383838] 合同履行期限:接到采购人通知后2日内供货完毕。 本项目不接受联合体投标。[/color][/size][/font][font=&][size=16px][color=#383838]二、申请人的资格要求:[/color][/size][/font][font=&][size=16px][color=#383838] 1、满足《中华人民共和国政府采购法》第二十二条规定; 2、落实政府采购政策需满足的资格要求:根据《政府采购中小企业发展管理办法》第六条第三款“符合下列情形之一的,可不专门面向中小企业预留采购份额:(三)按照本办法规定预留采购份额无法确保充分供应、充分竞争,或者存在可能影响政府采购目标实现的情形;”之规定,本项目不专门面向中小企业预留采购份额; 3、本项目的特定资格要求:无[/color][/size][/font][font=&][size=16px][color=#383838]三、获取采购文件:[/color][/size][/font][font=&][size=16px][color=#383838] 1.时间:2023年8月4日17时30分至2023年8月14日15时0分,每天上午00:00至11:59,下午12:00至23:59(北京时间,法定节假日除外 ) 2.地点:中国山东政府采购网(www.ccgp-shandong.gov.cn)或济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang) 3.方式:自行下载 4.售价:0[/color][/size][/font][font=&][size=16px][color=#383838]四、响应文件提交:[/color][/size][/font][font=&][size=16px][color=#383838] 1.截止时间:2023年8月15日9时30分(北京时间) 2.地 点:所有响应文件应于2023年8月15日9时30分(北京时间)之前上传到济宁市公共资源交易服务中心汶上分中心网,未在规定时间内上传响应文件电子文档或截止时间后上传的电子文档将不予公开唱标及评审。[/color][/size][/font][font=&][size=16px][color=#383838]五、开启:[/color][/size][/font][font=&][size=16px][color=#383838] 1.开启时间:2023年8月15日9时30分(北京时间) 2.开启地点:登录济宁市公共资源交易网不见面开标大厅进行解密[/color][/size][/font][font=&][size=16px][color=#383838]六、公告期限:[/color][/size][/font][font=&][size=16px][color=#383838] 自本公告发布之日起3个工作日。[/color][/size][/font][font=&][size=16px][color=#383838]七、其他补充事宜:[/color][/size][/font][font=&][size=16px][color=#383838] 其他补充事宜:1.项目参与:如潜在供应商予参与本项目,须同时在中国山东政府采购网(www.ccgp-shandong.gov.cn)、济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)登录、注册,响应本项目。未按要求响应本项目的,按无效报价处理。2.具体响应如下:(1)济宁市公共资源交易服务中心汶上分中心网(http://jnggzy.jnzbtb.cn/WenShang)—用户登录——政府采购,响应本项目并下载文件。【凡未在济宁市公共资源交易服务中心汶上分中心注册的供应商应先办理注册(济宁市公共资源交易服务中心汶上分中心网站-用户注册-政府采购)。所有项目全流程电子化并无任何纸质文件,所以注册后必须办理CA及电子签章,CA办理及系统操作咨询:0537-7817075、0537-6050123。已办理过注册及电子签章的供应商,登录系统,参与本项目。】(2)中国山东政府采购网(www.ccgp-shandong.gov.cn)-用户登录点击报名,报名后没有其他操作,无须在该网站上传电子版响应文件及报价。供应商未注册的先进行注册。(3)济宁市公共资源交易服务中心汶上分中心网与济宁市公共资源交易服务中心网在注册,登录、响应项目操作过程是通用的。各供应商在投标前须办理企业网上注册手续,具体程序详见济宁市公共资源交易服务中心《政府采购注册流程》。3.电子版文件上传1)本项目实行网上报价和上传响应文件电子文档所有响应文件应于2023年8月15日9时30分(北京时间)之前上传到济宁市公共资源交易服务中心汶上分中心网,未在规定时间内上传响应文件电子文档或截止时间后上传的电子文档将不予公开唱标及评审。2)具体操作:登录“济宁市公共资源交易服务中心汶上分中心网”用户登录政府政府采购,点击项目,上传文件。4.本项目公告将同时在济宁市公共资源交易服务中心汶上分中心网、中国山东政府采购网发布。本项目如有必要澄清和修改需要发布更正公告(变更公告)的,将同时在济宁市公共资源交易服务中心汶上分中心网、中国山东政府采购网及时发布,请各潜在供应商及时关注相关信息。更正公告(变更公告)一旦发布即视为通知所有潜在供应商。5.本项目为资格后审。6.本项目共分为2个包组。按包组先后顺序确定拟成交包组,开标、评标顺序按照项目包组顺序。包组开标顺序:A包、B包依次开标。包组评审顺序:A包、B包依次评审。请参与响应的企业认真核对所响应包组,避免上传文件出现失误。[/color][/size][/font][font=&][size=16px][color=#383838]八、对本次招标提出询问,请按以下方式联系:[/color][/size][/font][font=&][size=16px][color=#383838] 1、采购人信息 名 称:[/color][/size][/font][font=&][size=16px][color=#383838]汶上县检验检测中心 地 址:(汶上县检验检测中心) 联系方式:1668805347 2、采购代理机构 名 称:[/color][/size][/font][font=&][size=16px][color=#383838]山东政联建设项目管理有限公司 地 址:山东省济宁市高新区县(区)洸河百丰商贸中心号十九层 联系方式:15563772976 3、项目联系方式 项目联系人:孔芬 联系方式:15563772976[/color][/size][/font]
新华网北京4月20日电(记者吕诺、吴晶)正在公开征求意见的新《药品注册管理办法》征求意见稿提出,国家食品药品监管局根据 申请人提供的研究数据,对药品的安全性、有效性和质量可控性进行系统评价,对上市价值和风险进行评估,在此基础上决定是否同意该药品上市。 国家食品药品监管局副局长吴浈20日对此评价说,评估是指药品监督管理部门站在公众利益的角度,综合技术审评、国家药品政策、环境保护、药物经济等,对一类药品或某种药品进行综合评价,以确定是否批准上市的行为。这种评估是根据需要,由有关专家、相关部门组成的评价委员会进行的。这一原则的设定,旨在为应对可能出现的非理性申报奠定法规基础。 吴浈说,我国现行的《药品注册管理办法》,存在审评审批权力配置不合理,标准不够完善,程序不够严密,过程不够透明,审评审批时限达不到要求等突出问题。征求意见稿在药品审评审批环节进行了多处修订,以期推动药品审评审批改革。 据吴浈介绍,征求意见稿调整和明确了监督管理部门的职责,强化了省级药监部门在对研制情况和申报资料真实性核查方面的责任;增加了省级药监局对申报资料进行初步审查,并提出审查意见的权利,以及组织实施仿制药、补充申请注册的批准生产前的现场生产检查工作的责任。国家食品药品监管局还将探索将部分审评或审批事项下放给省局的方法。 为加强对审评审批权力的约束,征求意见稿还提出实施公示制度,公示药品注册受理、核查、检验、审评、审批人员名单及相关信息,公示药品注册的检验、审评、审批进展情况,公示药品审评审批结果,接受社会监督。
[quote]项目概况济南市食品药品检验检测中心试剂耗材采购项目 采购项目的潜在供应商应在济南市历下区二环东路5001号和瑞广场A座8层获取采购文件,并于2023年10月13日 14点00分(北京时间)前提交响应文件。[/quote][font=inherit]一、项目基本情况[/font]项目编号:SDITC-7-2023-169项目名称:济南市食品药品检验检测中心试剂耗材采购项目采购方式:竞争性磋商预算金额:50.0000000 万元(人民币)最高限价(如有):50.0000000 万元(人民币)采购需求:详见磋商文件合同履行期限:详见磋商文件本项目( 不接受 )联合体投标。[font=inherit]二、申请人的资格要求:[/font]1.满足《中华人民共和国政府采购法》第二十二条规定;2.落实政府采购政策需满足的资格要求:详见磋商文件3.本项目的特定资格要求:1、符合《中华人民共和国政府采购法》第二十二条的规定;2、在中国境内注册,具有独立法人资格;3、危险化学品经营许可证(其中包括易制爆类危化品经营范围),非药品类易制毒化学品经营备案证明等;4、本项目不接受联合体报价。[font=inherit]三、获取采购文件[/font]时间:2023年09月22日 至 2023年09月28日,每天上午9:00至11:30,下午13:00至17:00。(北京时间,法定节假日除外)地点:济南市历下区二环东路5001号和瑞广场A座8层方式:现场报名时须提供以下证件加盖公章的复印件一套:①营业执照副本、②法定代表人授权委托书及被授权人身份证、③危险化学品经营许可证(其中包括易制爆类危化品经营范围),非药品类易制毒化学品经营备案证明等。售价:¥400.0 元(人民币)[font=inherit]四、响应文件提交[/font]截止时间:2023年10月13日 14点00分(北京时间)地点:济南市历下区二环东路5001号和瑞广场A座8层开标室[font=inherit]五、开启[/font]时间:2023年10月13日 14点00分(北京时间)地点:济南市历下区二环东路5001号和瑞广场A座8层开标室[font=inherit]六、公告期限[/font]自本公告发布之日起3个工作日。[font=inherit]七、其他补充事宜[/font][font=inherit]八、凡对本次采购提出询问,请按以下方式联系。[/font]1.采购人信息名 称:济南市食品药品检验检测中心地址:济南高新区港兴三路北段济南药谷4号楼联系方式:秦老师 0531-55515931 130827539572.采购代理机构信息名 称:山东省国际招标有限公司地 址:济南市历下区二环东路5001号和瑞广场A座8层联系方式:赵吉松/贾式一0531-88193755转8037/80683.项目联系方式项目联系人:赵吉松/贾式一电 话: 0531-88193755转8037/8068
药品生产企业GMP工作手册 1、药品生产质量管理规范(98年版) 附录2、粉针剂实施《药品生产质量管理规范》指南3、粉针剂实施《药品生产质量管理规范》检查评审细则4、大容量注射剂实施《药品生产质量管理规范》指南5、大容量注射剂实施《药品生产质量管理规范》检查评审细则6、原料药实施《药品生产质量管理规范》指南7、原料药实施《药品生产质量管理规范》检查评审细则8、片剂、硬胶囊剂、颗粒剂实施《药品生产质量管理规范》指南9、片剂、硬胶囊剂、颗粒剂实施《药品生产质量管理规范》检查评审细则10、小容量注射剂实施《药品生产质量管理规范》指南11、小容量注射剂实施《药品生产质量管理规范》检查评审细则12、关于医药行业组织实施GMP认证工作有关问题的通知13、关于印发医药行业实施GMP“九五”计划和医药行业实施GSP“九五”计划的通知14、关于印发《药品生产企业(车间)实施GMP达标验收管理办法》的通知15、关于印发《药品生产企业(车间)实施GMP现场检查评审工作程序》的通知16、关于药品生产企业(车间)GMP达标验收工作有关规定的通知17、关于发布《医药工业洁净厂房设计规范》的通知 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=46811]药品生产企业GMP工作手册[/url]
[quote][b]项目概况[/b]2023年重庆海关技术中心试剂药品耗材采购项目 招标项目的潜在投标人应在重庆市渝北区黄山大道两江星界2栋17-1获取招标文件,并于2023年05月05日 10点00分(北京时间)前递交投标文件。[/quote][font=inherit]一、项目基本情况[/font]项目编号:HCCQZB-2023-10项目名称:2023年重庆海关技术中心试剂药品耗材采购项目预算金额:200.0000000 万元(人民币)采购需求:/合同履行期限:自合同签订之日起三年本项目( 不接受 )联合体投标。[font=inherit]二、申请人的资格要求:[/font]1.满足《中华人民共和国政府采购法》第二十二条规定;2.落实政府采购政策需满足的资格要求:落实政府采购政策需满足的资格要求:落实中小微企业、监狱企业、残疾人福利性单位采购优惠政策。3.本项目的特定资格要求:(三)特殊资格要求:(1)具备危险化学品经营、气瓶充装资质;具备危险品道路运输资质或与具备危险品道路运输资质的单位签订委托运输协议。(竞选气体产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中序号1645-1655)。(2)非药品类易制毒化学品经营备案证明(竞选易制毒产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中1656-1661)。(3)易制爆危险化学品从业单位备案证明(竞选易制爆产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中1662-1664)。[font=inherit]三、获取招标文件[/font]时间:2023年04月07日 至 2023年04月14日,每天上午8:00至14:00,下午12:00至21:00。(北京时间,法定节假日除外)地点:重庆市渝北区黄山大道两江星界2栋17-1方式:采取线下发售方式。投标人携带“投标人资格要求”的资料复印件加盖企业鲜章,赴招标代理机构处(重庆市渝北区黄山大道两江星界2栋17-1),经审查合格后领取招标文件。售价:¥500.0 元,本公告包含的招标文件售价总和[font=inherit]四、提交投标文件截止时间、开标时间和地点[/font]提交投标文件截止时间:2023年05月05日 10点00分(北京时间)开标时间:2023年05月05日 10点00分(北京时间)地点:重庆市渝北区黄山大道两江星界2栋17-1[font=inherit]五、公告期限[/font]自本公告发布之日起5个工作日。[font=inherit]六、其他补充事宜[/font]红城国际工程项目管理有限公司(以下简称:采购代理机构)受重庆海关技术中心(以下简称:采购人)的委托,对2023年重庆海关技术中心试剂药品耗材采购项目进行公开招标,欢迎有资格的投标人参加投标。[b][font=inherit]一、招标项目内容[/font][/b][table][tr][td][align=center][font=inherit]项目名称[/font][/align][/td][td][align=center][font=inherit]最高限价[/font][/align][align=center][font=inherit](万元)[/font][/align][/td][td][align=center][font=inherit]投标保证金[/font][/align][align=center][font=inherit](万元)[/font][/align][/td][td][align=center][font=inherit]中标人数量(名)[/font][/align][/td][td][align=center][font=inherit]采购标的对应的中小企业划分标准所属行业[/font][/align][/td][/tr][tr][td]2023年重庆海关技术中心试剂药品耗材采购项目[/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][td][align=center]其他未列明行业[/align][/td][/tr][/table][b][font=inherit]二、资金来源[/font]其他资金及财政拨款资金。[font=inherit]三、履行期限[/font][/b]合同履行期限:自合同签订之日起三年。合同每年签订一次,续签前采购人对供应商服务进行评价,如评价不合格,则取消续签。续签时采购人和供应商协商一致后,均可根据试剂药品耗材的市场价格对首次合同内的采购品目进行调减。[b][font=inherit]四、投标人资格要求[/font][/b](一)满足《中华人民共和国政府采购法》第二十二条规定。(二)落实政府采购政策需满足的资格要求:落实中小微企业、监狱企业、残疾人福利性单位采购优惠政策。(三)特殊资格要求:(1)具备危险化学品经营、气瓶充装资质;具备危险品道路运输资质或与具备危险品道路运输资质的单位签订委托运输协议。(竞选气体产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中序号1645-1655)。(2)非药品类易制毒化学品经营备案证明(竞选易制毒产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中1656-1661)。(3)易制爆危险化学品从业单位备案证明(竞选易制爆产品提供,详见《2023年重庆海关技术中心试剂药品耗材采购遴选目录》中1662-1664)。[b][font=inherit]五、投标、开标有关说明[/font][/b](一)凡有意参加投标的投标人,请到采购代理机构领取本项目招标文件以及清单、澄清等开标前公布的所有项目资料,无论投标人领取与否,均视为已知晓所有招标内容。(二)招标文件公告期限:自采购公告发布之日起五个工作日。(三)招标文件提供期限(1)招标文件提供期限:2023年04月07日至2023年04月14日。(2)报名方式:1.采取网上发售方式。投标人采取发送电子邮件方式递交报名资料,邮件主题:项目名称+项目编号式;邮件附件:需采用A4纸幅面,将“投标人资格要求”的材料加盖企业鲜章的扫描件,按顺序制作成一套资料;邮件内容:列明公司名称、法定代表人或授权代表人姓名及联系方式的PDF格式文件,文件名称与主题一致,复印件扫描无效。报名材料审核通过后,招标代理机构联系人向供应商邮箱发送招标文件电子版;审核未通过的,招标代理机构联系人以邮件形式回复审核情况,供应商可在招标文件申领时间内重新提交材料。招标代理机构邮箱2533086645@qq.com。2.采取线下发售方式。投标人携带“投标人资格要求”的资料复印件加盖企业鲜章,赴招标代理机构处(重庆市渝北区黄山大道两江星界2栋17-1),经审查合格后领取招标文件。(3)招标文件售价:人民币500.00元/包。(报名资料审核通过后会将收款码发送至报名单位。)(四)投标地点:重庆市渝北区黄山大道两江星界2栋17-1。(五)投标截止时间:2023年05月05日北京时间10:00。(六)开标时间:2023年05月05日北京时间10:00。(七)开标地点:同投标地点。[b][font=inherit]六、采购项目需落实的政府采购政策[/font][/b](一)按照《财政部 生态环境部关于印发环境标志产品政府采购品目清单的通知》(财库〔2019〕18号)和《财政部 发展改革委关于印发节能产品政府采购品目清单的通知》(财库〔2019〕19号)的规定,落实国家节能环保政策。(二)按照财政部、工业和信息化部关于印发《政府采购促进中小企业发展管理办法》的通知(财库〔2020〕46号)的规定,落实促进中小企业发展政策。(三)按照《财政部、司法部关于政府采购支持监狱企业发展有关问题的通知》(财库〔2014〕68号)的规定,落实支持监狱企业发展政策。(四)按照《三部门联合发布关于促进残疾人就业政府采购政策的通知》(财库〔2017〕 141号)的规定,落实支持残疾人福利性单位发展政策。[b][font=inherit]七、投标有关规定[/font][/b](一)单位负责人为同一人或者存在直接控股、管理关系的不同投标人,不得参加同一合同项(包)下的政府采购活动。(二)本项目若有澄清文件一律在重庆海关官方网站、中国政府采购网、中国采购与招标网上发布,请各投标人注意下载或到采购代理机构领取;无论投标人下载或领取与否,均视同投标人已知晓本项目澄清文件的内容。(四)超过投标截止时间递交的投标文件,恕不接收。(五)投标费用:无论投标结果如何,投标人参与本项目投标的所有费用均应由投标人自行承担。[font=inherit](六)本项目不接受联合体参与投标,否则按无效投标处理。[/font][font=inherit](七)本项目不接受合同分包、转包,否则按无效投标处理。[/font](八)按照《财政部关于在政府采购活动中查询及使用信用记录有关问题的通知》(财库〔2016〕125号),投标人列入失信被执行人、重大税收违法案件当事人名单、政府采购严重违法失信行为记录名单及其他不符合《中华人民共和国政府采购法》第二十二条规定条件的投标人,将拒绝其参与政府采购活动。[b][font=inherit]八、联系方式[/font][/b](一)采购人:重庆海关技术中心联系人:冉老师电 话:023-67725389地 址:重庆市江北区红黄路8号(二)项目联系方式联系人:赵老师、柯老师电 话:023-67724310、023-677234750(三)采购代理机构: 红城国际工程项目管理有限公司联系人:刘老师电 话:023-67559507、15823887464地 址: 重庆市渝北区黄山大道两江星界2栋17-1[font=inherit]七、对本次招标提出询问,请按以下方式联系。[/font]1.采购人信息名 称:重庆海关技术中心地址:重庆市江北区红黄路8号联系方式:赵老师023-677243102.采购代理机构信息名 称:红城国际工程项目管理有限公司地 址:重庆市渝北区黄山大道两江星界2栋17-1联系方式:刘亚茜023-675595073.项目联系方式项目联系人:刘亚茜电 话: 023-67559507







 我要推广仪器
我要推广仪器
 下载APP
下载APP