“呼口气”就能检测癌症?嗯,临床试验进行中!
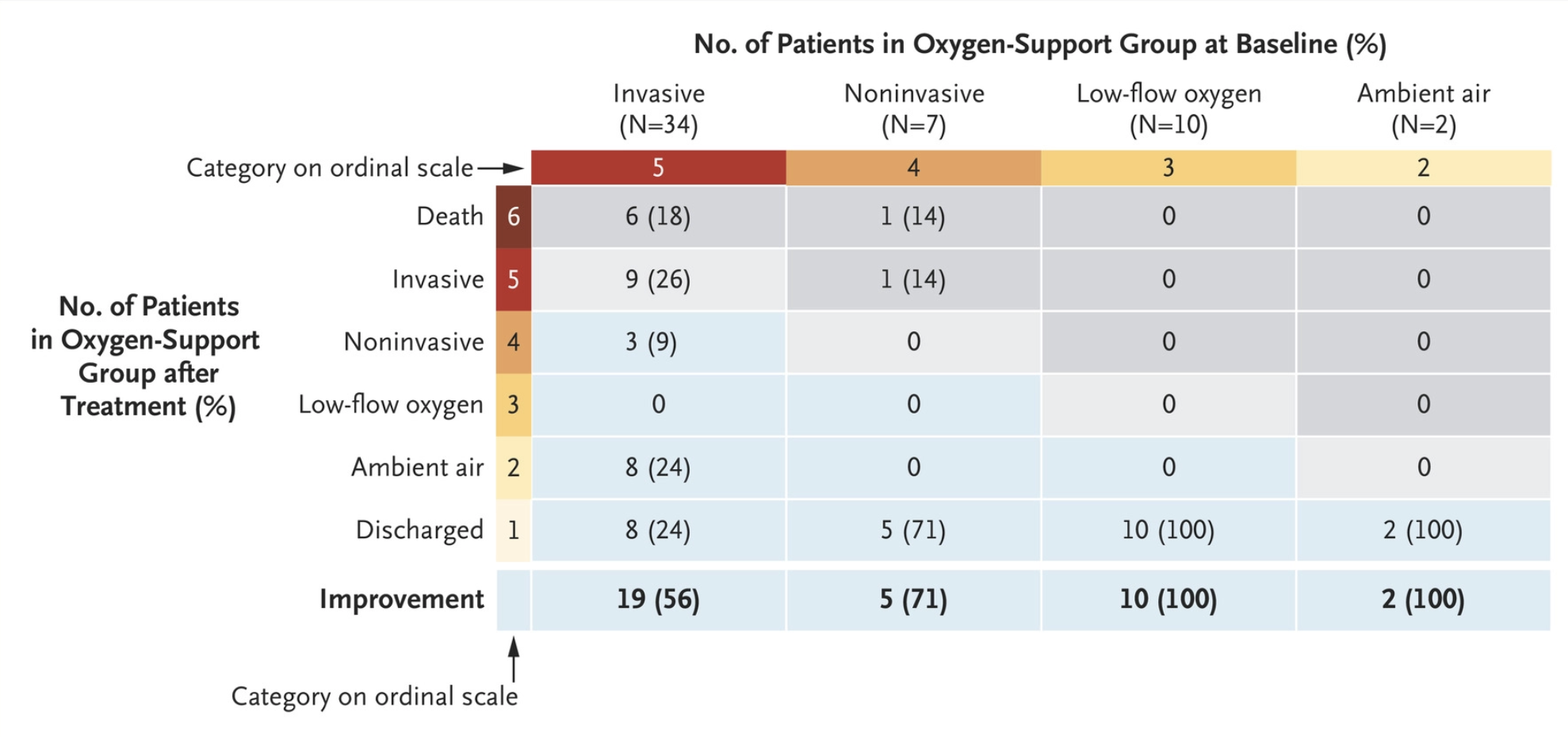
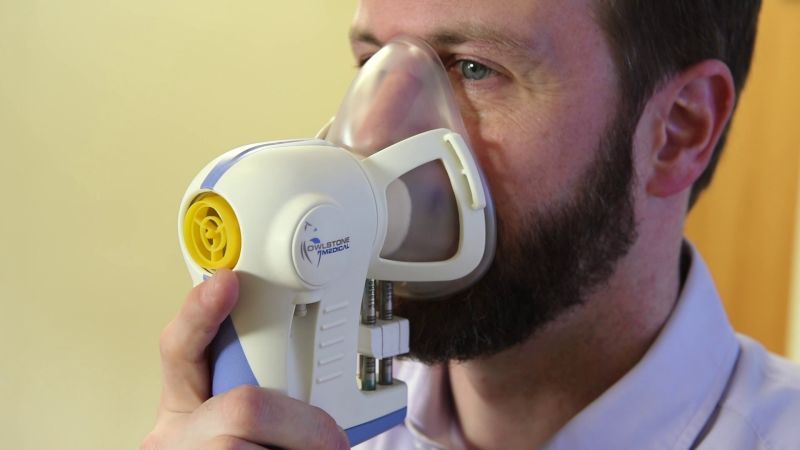
p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201901/uepic/b383d269-1964-4e37-ac77-666bbbc6c86a.jpg" title=" 1.jpg" alt=" 1.jpg" width=" 430" height=" 244" style=" width: 430px height: 244px " / /p p style=" text-indent: 2em " 用于检测多种癌症的呼吸测试正在进行临床试验。开发者们认为,这项新技术有望大大降低癌症诊断的成本。(图片来源:yahoo) /p p style=" text-indent: 2em " strong style=" color: rgb(0, 112, 192) text-align: center " “呼口气”就能检测癌症? /strong /p p 听起来如此“炫酷”的技术正在英国进行关键的临床试验。 /p p 慈善机构Cancer Research UK联合一家名为Owlstone Medical的公司希望通过该试验调查,“呼吸测癌”技术——Breath Biopsy& reg 用于检测多种癌症的可行性。 /p p 具体来说,试验将收集来自1500人的呼吸样本,从疑似食道癌和胃癌患者开始,然后在未来几个月扩大到前列腺癌、肾癌、膀胱癌、肝癌和胰腺癌患者中。此外,健康的人也将被纳入该试验。 /p p 在剑桥的Addenbrooke医院,参与者将被要求对着面罩呼吸10分钟,以便收集样本。之后,样本会被送往实验室进行分析。患者可以在几天内得到结果。 /p p style=" text-align: center " span style=" color: rgb(192, 0, 0) " strong 01“呼吸测癌”测什么 /strong /span /p p 事实上,“呼吸测癌”并不是一种新概念,其背后的科学原理也不新鲜。这种技术检测的主要是一类被称为“挥发性有机化合物”(volatile organic compounds,VOCs)的分子,如醛和酮。 /p p 当人体细胞进行生化反应时,VOCs这类分子会被释放出来。然而,如果人患了癌症或其它疾病,细胞的代谢模式会发生改变,进而释放不同模式和(或)数量的VOCs。 /p p 因安全无创、简单便捷、接受度高等特点,呼吸检测一直是疾病诊断领域研究的热点。先前,科学家们已经发表过一些针对胃癌、肺癌、前列腺癌、食道癌、结肠癌的呼吸测试技术。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201901/uepic/f95c324a-e1f2-4862-9b3d-f89ff8df92e0.jpg" title=" 2.png" alt=" 2.png" / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " 部分“呼吸测癌”相关的论文 /span /p p 举例来说,去年5月,同样是来自英国的科学家在JAMA Oncology杂志上报道了一种能够成功检测出食道癌和胃癌的呼吸测试。在一个包含335名患者的多中心临床试验中,这一无创的呼吸测试能够以85%的准确率从良性疾病中鉴定出癌症。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201901/uepic/346fc858-e145-4ee3-a0f8-21b55f7c5343.jpg" title=" 3.jpg" alt=" 3.jpg" / /p p span style=" color: rgb(127, 127, 127) " Hanna教授团队也将进行更大规模的临床试验以验证现有的结果,并看看该测试能否诊断其它类型的癌症,如胰腺癌。(Credit: Imperial College London) /span /p p 领导这一研究的 George Hanna教授说:“胃癌和食道癌大多数是在晚期才被诊断出来,而此时想要治愈是不可能的。因此,癌症的早期诊断是一种刚需,能够给患者带来更多的治疗选择,挽救更多的生命。” /p p style=" text-align: center " strong span style=" color: rgb(192, 0, 0) " 02 彻底改变癌症检测 /span /strong /p p Billy Boyle在剑桥大学的TED演讲: strong a href=" https://v.qq.com/x/page/u08230obphv.html" target=" _blank" 一个简单的呼吸测试能诊断疾病吗? /a /strong /p p 与针对一种或少数癌症的呼吸测试技术不同,Owlstone Medical公司的Breath Biopsy& reg 技术是一种“通用型呼吸检测”,有望用于检测多种不同类型的癌症。 /p p 基于此次开展的临床试验,科学家们想要弄清楚,是否不同癌症类型的代谢特征是相似或不同的,以及这项技术能够多早发现其中任何的异常特征。 /p p 如果这项技术最终被证实可用,开发者们希望,它可以作为癌症检测的“第一步”筛查,以判定“被检测者”是否需要进行其它检测,与血液和尿液检测一起,帮助医生实现癌症早期诊断。 /p p Cancer Research UK的David Crosby博士认为,包括Breath Biopsy& reg 在内的呼吸测试技术有可能彻底改变未来检测和诊断癌症的方式。此外,有医生表示,这类技术可能会在10年内最终取代目前的癌症筛查手段。 /p p “我们迫切需要开发新的工具,比如这种呼吸测试,它可以帮助我们更早地发现和诊断癌症,为患者提供最佳的生存机会。”领导该临床试验的Rebecca Fitzgerald教授说。 /p p style=" text-align: center " strong span style=" color: rgb(192, 0, 0) " 03 背后的感人故事 /span /strong /p p 查阅资料时,笔者注意到,Breath Biopsy& reg 这一技术开发的背后有着非常触动人心的故事。 /p p 2014年圣诞节,癌症夺走了Owlstone Medical公司联合创始人兼CEO Billy Boyle妻子Kate Gross的生命。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201901/uepic/54964ccb-9e85-4b75-9b1f-b6239df8b65a.jpg" title=" 4.jpg" alt=" 4.jpg" / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " Gross女士 /span /p p Gross女士的一生虽短暂,但不平凡。她曾在牛津大学学习英语,工作后担任过英国两位首相的顾问。去世那一年,她还因在非洲的慈善工作被授予大英帝国勋章之官佐勋章(OBE)。 /p p 2012年10月,Gross女士在一次出差途中感觉胃痛。从加州飞回家后,她直接去了医院。很快,医生确诊她患上了结肠癌,并且癌症已经扩散到了肝脏。2013年,在成功进行了肝脏肿瘤切除手术后,Gross女士能够继续活着的渺茫希望似乎变大了一些。 /p p 遗憾的是,不久后,她的癌症又复发了,并扩散到了骨头。在她与癌症抗争的过程中,化疗并没有起到多大帮助,2014年年底,药物彻底失效了。不久后,她的生命停在了36岁,留下其丈夫Billy Boyle独自陪伴他们的双胞胎儿子。 /p p style=" text-align: center" img src=" https://img1.17img.cn/17img/images/201901/uepic/08762673-3ae7-4dab-af9f-203105bdde4a.jpg" title=" 5.jpg" alt=" 5.jpg" / /p p style=" text-align: center " span style=" color: rgb(127, 127, 127) " Billy Boyle及其妻子和双胞胎儿子。 /span /p p 妻子的离开让Billy Boyle感受到了癌症对家庭巨大的破坏力。 /p p 尽管悲伤,但这位剑桥大学的工程学毕业生并没有因此一蹶不振,相反,他与人合伙创办了一家公司,也就是Owlstone Medical,希望彻底改变癌症的诊断。 /p p 因为,在他看来,如果能够早点诊断出癌症,他的家庭也许就不会遭受这样的厄运了。 /p p Billy Boyle希望,包括Breath Biopsy& reg 在内的一些新技术能够更早地检测出癌症,避免更多的家庭重复他们的悲伤故事。 /p p style=" text-align: center " span style=" color: rgb(192, 0, 0) " strong 04 写在最后 /strong /span /p p 2013年4月,发表在PLOS ONE上的一项研究表明,人类在呼吸时呼出的化合物其实像指纹一样独一无二。或许,在不久的将来,临床医生会像使用常规的血液检测一样运用呼吸测试。 /p





















 我要推广仪器
我要推广仪器
 下载APP
下载APP