新型冠状病毒科研进展之——蛋白靶点结构研究进展
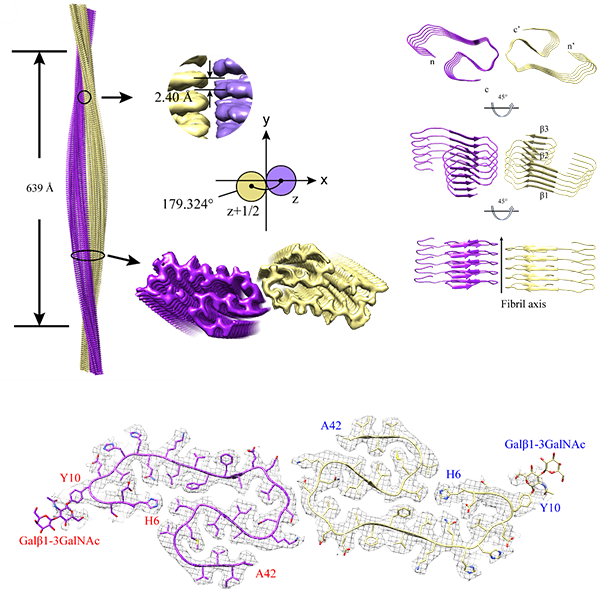
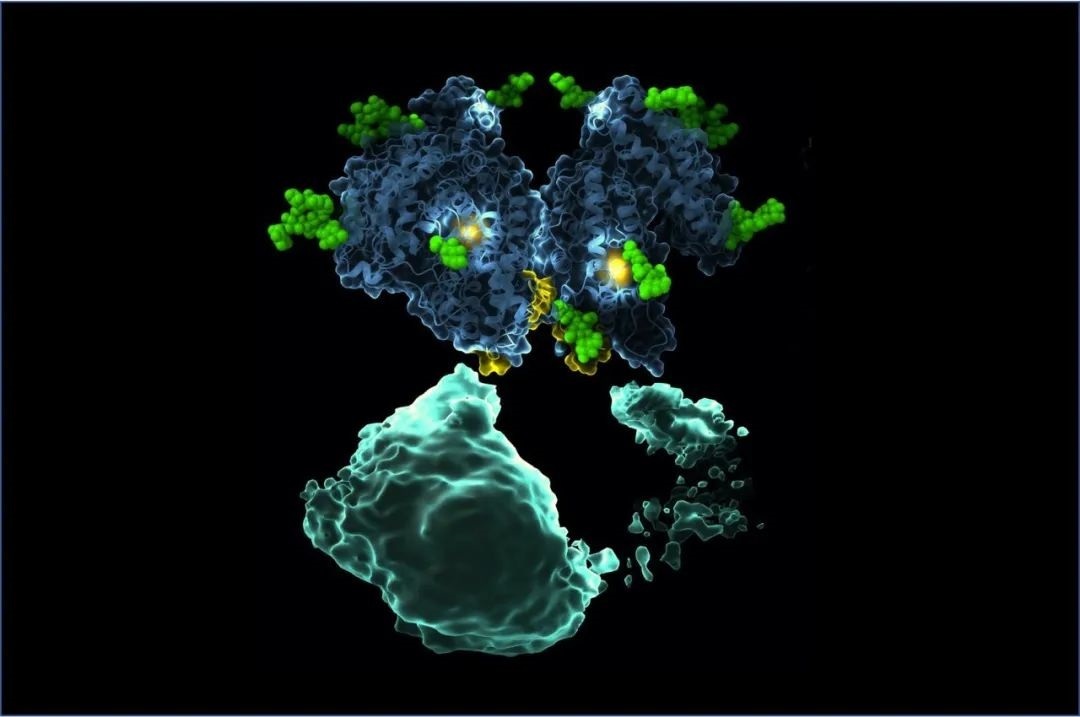
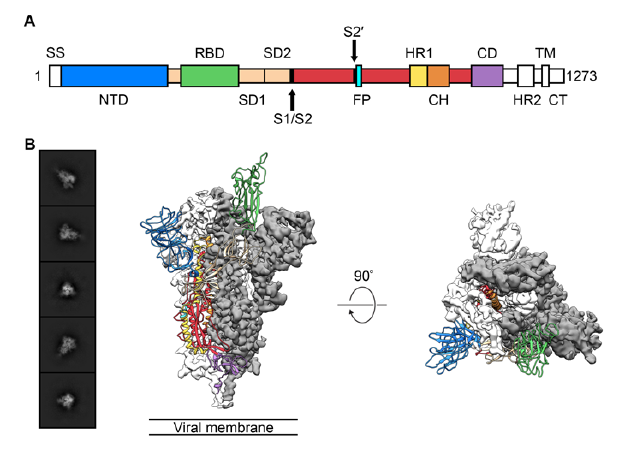
p style=" text-align: justify text-indent: 2em line-height: 1.75em " strong 仪器信息网讯& nbsp /strong span style=" text-indent: 2em " 冠状病毒是一类严重危害人类和动物健康的病原微生物,属于具有大量天然宿主的一类RNA病毒。该病毒极易发生基因重组和变异,具有遗传多样性,迄今为止,已不断有新亚型或新的冠状病毒出现。冠状病毒上的S蛋白、PLpro和3CLpro是药物开发的良好靶点,本文整理并总结了基于靶标发现的潜在药物。 /span /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " span style=" text-indent: 2em " /span /p p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202003/uepic/38dd544f-4905-4c6c-899f-7d2e0b1a4099.jpg" title=" 截屏2020-03-30上午11.54.47.png" alt=" 截屏2020-03-30上午11.54.47.png" / /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 冠状病毒是一种有包膜的、非节段的单股正链RNA病毒,属于巢病毒目(nidovirales)冠状病毒科(Coronaviridae)正冠状病毒亚科(ortho-coronavirinae)。由于病毒包膜上有向四周伸出的突起,形如花冠而得名。冠状病毒亚科进一步细分为四类,即α、β、γ 和 δ 冠状病毒。冠状病毒在自然界中广泛存在,其自然宿主包括人类和其他哺乳动物如牛、猪、犬、猫、鼠和蝙蝠等。 strong 目前,已经鉴定出六种人类冠状病毒,其中包括α属的HCoV-29E和HCoV-NL63;β属的HCoV-OC43、HCoV-HKU1、严重急性呼吸综合征相关冠状病毒(SARS-CoV)和中东呼吸综合征相关冠状病毒(MERS-CoV)。 /strong /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 另外,近期从武汉市不明原因肺炎患者下呼吸道分离出的冠状病毒,世界卫生组织初步命名为2019-nCoV。2020年2月12日,国际病毒分类委员会宣布新型冠状病毒(2019-nCoV)的正式分类名为 span style=" color: rgb(192, 0, 0) " 严重急性呼吸综合征冠状病毒(SARS-CoV-2) /span 。研究者将来源于武汉的新型冠状病毒序列与已知的“SARS冠状病毒”“MERS冠状病毒”进行了比较,发现 strong 6个新型冠状病毒序列几乎一致,其与SARS的同源性更高,相似性约为70%,与MERS相似性约为40%。 /strong strong 序列差异主要在ORF1a和编码S-蛋白的spike基因上,这是冠状病毒与宿主细胞作用的关键蛋白。 /strong /p p style=" text-align: center text-indent: 2em line-height: 1.75em " strong span style=" color: rgb(0, 112, 192) " 冠状病毒蛋白靶点结构研究进展 /span /strong /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 冠状病毒是最大的一种核糖核酸病毒(26~32kb),其基因组为单股、正链RNA。编码非结构蛋白(Nps)的复制酶基因占据了基因组的三分之二,而结构蛋白和辅助蛋白仅占病毒基因组的三分之一。目前已经解析出了许多冠状病毒相关的蛋白质结构,如SARS-CoV S糖蛋白(PDB ID:5WRG)(图1A)、MERS-CoV N蛋白的C末端结构域(PDB ID:6G13)(图1B)、MERS-CoV N蛋白的N末端结构域(PDB ID: 4UD1)(图1C)。 /p p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202003/uepic/fd7a83a8-25f3-48a0-989e-958bb95bc364.jpg" title=" 截屏2020-03-30上午10.38.54.png" alt=" 截屏2020-03-30上午10.38.54.png" / /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 病毒体与宿主细胞的初始附着是通过S蛋白与其受体之间的相互作用而开始的。根据研究报道,S蛋白具有受体结合活性和膜融合活性,是冠状病毒感染细胞的关键蛋白。研究发现在大多数冠状病毒中,S蛋白被宿主细胞弗林蛋白酶(Furin)样蛋白酶切割成S1和S2两种单独的多肽。S1的主要功能是与宿主细胞表面受体结合,而S2亚基则负责介导病毒-细胞以及细胞-细胞膜的融合。 /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 在对近期的SARS-CoV-2 S蛋白进行研究时发现,虽然SARS-CoV-2 S蛋白中与ACE2蛋白结合的5个关键氨基酸中有4个发生了变化,但变化后的氨基酸,却没有影响SARS-CoV S蛋白与ACE2 蛋白互作的构象。与SARS-CoV S蛋白相比,突变体后的SARS-CoV-2 S蛋白结构与ACE2 蛋白相互作用能力,由于丢失的少数氢键有所下降,但仍然达到很强的结合自由能,说明SARS-CoV-2 是通过S蛋白与人ACE2相互作用感染人的呼吸道上皮细胞。 /p p style=" text-align: center text-indent: 2em line-height: 1.75em " span style=" color: rgb(0, 112, 192) " strong 疫苗和治疗药物研究进展 /strong /span /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 为了控制病毒的爆发,研究者们开发了针对 SARS¯ CoV 和 MERS¯ CoV 的疫苗。不同的疫苗有不同的制备方法下表中列出了这些方法的发展和优缺点。 /p p style=" text-align: center" img style=" max-width:100% max-height:100% " src=" https://img1.17img.cn/17img/images/202003/uepic/446bbee2-3399-4764-a3f5-0339be331bc9.jpg" title=" 截屏2020-03-30上午10.59.39.png" alt=" 截屏2020-03-30上午10.59.39.png" / /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 迄今为止,大多数研究只集中在SARS疫苗的开发上,研究过程中使用了动物模型,但是这些模型并不能概括人类发生的严重临床疾病。 strong 综合SARS 和 MERS 疫苗的研究经验。发现冠状病毒疫苗的研究主要靶标是冠状病毒的S蛋白。疫苗不仅需要诱导体液和细胞免疫应答,还需要诱导黏膜免疫应答并借助佐剂来诱导 Th1 和 Th2 途径的平衡。也就是说成功的疫苗必须在不引起过度免疫激活的情况下达到保护的平衡。 未来还需加强对 SARS-CoV 和 MERS-CoV 等疫苗的研发。 /strong /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 对于目前的 SARS-CoV-2,据新华社报道,美国医学专家正与中国同行合作研发针对新型冠状病毒的疫苗,美国休斯敦贝勒医学院彼得霍特兹教授通过电子邮件表示,贝勒医学院正在与美国得克萨斯大学、美国纽约血液中心以及中国上海复旦大学合作开发疫苗。目前,尚无针对 SARS-CoV、MERS-CoV、 & nbsp SARS-CoV-2 和其他 HCoV 感染的特异性疗法,患者主要接受支持性治疗,并辅以多种药物组合,包括使用抗体、干扰素以及病毒和宿主蛋白酶的抑制剂。& nbsp /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 此外,除了针对SARS外,有研究报道了一种针对MERS-CoV S蛋白N端结构域的新型中和单克隆抗体。该研究表明N末端结构域在病毒感染过程中可能很重要,这项发现对于进一步的疫苗设计和针对MERS-CoV感染的预防和治疗性单克隆免疫法的开发具有重要意义。 /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 理想情况下,疫苗接种和抗病毒治疗都应具有各自明确的作用机制,以避免产生逃逸突变病毒菌株,并提高对不同病毒菌株的活性。 strong 迄今为止,利巴韦林和利巴韦林加各种类型的干扰素已成为SARS和MERS患者最常用的治疗手段。 /strong SARS-CoV-2爆发以来,全国各个攻关团队筛选出一系列具有治疗潜力的药物。 strong 中国科学院上海药物研究所和上海科技大学免疫化学研究所的抗SARS-CoV-2病毒感染联合应急攻关团队报道了综合利用虚拟筛选和酶学测试相结合的策略进行药物筛选,发现了30种可能对SARS-CoV-2有治疗作用的药物、活性天然产物和中药。 /strong /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " strong span style=" text-indent: 2em " 沈阳药科大学、华中科技大学和军事医学研究院国家应急防控药物工程技术研究中心组成的联合攻关小组发现SARS-CoV-2蛋白序列中SARS-CoV-2-PLP序列与SARS-CoV-PLP具有82%的氨基酸同源性。 /span /strong /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " & nbsp 2020 年 1 月 21 日,中国科学院上海巴斯德研究所郝沛研究员等使用计算机模拟的方法发现了& nbsp SARS-CoV-2的S-蛋白的受体结合结构域(RBD)和人血管紧张素转化酶 ACE2 的结合作用较强。& nbsp SARS-CoV-2通过 S 蛋白 - ACE2 结合途径对人 类传播构成了重大的公共卫生风险。因此ACE2 也可能用于& nbsp SARS-CoV-2的治疗研究。 黄朝林等根据过往洛匹那韦利托那韦片对& nbsp SARS-CoV感染的患者有“ 实质性的临床益处” 的结果 推测这种疗法可能对& nbsp SARS-CoV-2感染的患者有效。此外,武汉病毒研究所与军事医学科学院毒物药物研究所联合发现了在细胞层面上对& nbsp SARS-CoV-2有较好抑 制作用的雷米迪维或瑞德西韦(RemdesivirGS-5734)、氯喹(ChloroquineSigma-C6628)、利托那 韦(Ritonavir)等三种“老药物”。& nbsp /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 瑞德西韦属于核苷类似物能够抑制 RNA 依赖的 RNA 聚合酶 (RdRp),由美国知名药企吉利德科学公司研发原本用于对抗埃博拉病毒在体外和动物模型中瑞德西韦证实了对 SARS 和 MERS 的病毒病原体均有活性它们与新型冠状病毒结构相似,从理论预测瑞德西韦对新型冠状病毒可能有效。 /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 目 strong 前瑞德西韦已进入 III 期临床试验该临床试验项目将在武汉市金银潭医院等多家医院同时进行两部分组成均采用随机、双盲、安慰剂对照形式开展。 /strong 据吉利德对外披露,在武汉进行的临床实验有两项,一是研究评估瑞德西韦用于未表现出显著临床症状患者的治疗效果,也就是轻、重症患者。另一项则是评估其用于重症确诊病患的疗效。值得一提的是,来自中国科学院武汉病毒研究所等机构的中国学者已经在细胞水平上验证了瑞德西韦在2019 新型冠状病毒上有较好的活性。 span style=" text-indent: 2em " 研究结果显示在 Vero E6 细胞上瑞德西韦对 SARS-CoV-2的半数有效浓度EC50 =0.77μmol/L,选择指数 SI 大于 129,表明该药物在细胞水平上能效抑制& nbsp SARS-CoV-2 的感染,但其在人体上的作用还有待临床验证。 /span /p p br/ /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 参考文献: /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 1.XU X T,CHEN P,WANG J F,et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission[J]. Science China-Life Sciences,2020.& nbsp /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 2.FOUCHIER R A,HARTWIG N G,BESTEBROER T M,et al. A previously undescribed coronavirus associated with respiratory disease in humans [J]. Proceedings of the National Academy of Sciences of the United States of America,2004,101(16):6212 - 6216.& nbsp /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 3.VANDER HOEK L,PYRC K,JEBBINK M F,et al. Identification of a new human coronavirus [ J] . Nature Medicine,2004,10(4):368 -373.& nbsp /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 4.WANG M,CAO R,ZHANG L,et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019¯ nCoV) in vitro [J]. Cell Research,2020 /p p style=" text-align: justify text-indent: 2em line-height: 1.75em " 5.HUANG C,WANG Y,LI X,et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan,China [J]. Lancet,2020.& nbsp /p p br/ /p p br/ /p





















 我要推广仪器
我要推广仪器
 下载APP
下载APP