“雕刻”细胞!浙大、北大联手在单细胞蛋白质组学分析研究领域取得新突破!
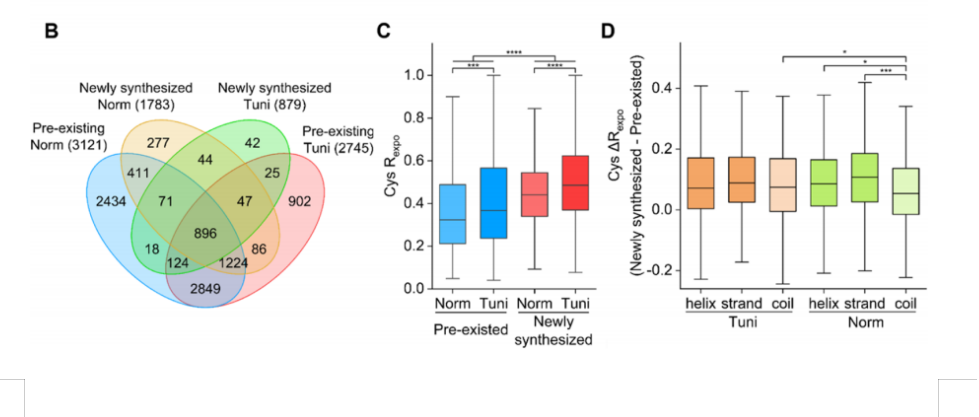
p span style=" color: rgb(0, 112, 192) " 近日,浙江大学方群团队联合北京大学黄超兰团队在单细胞蛋白质组学分析研究领域取得了突破性进展。 /span 此项成果近日以全文形式在线发表于美国化学会的Analytical Chemistry杂志上(影响因子 6.32),论文题目为“Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis”。 /p p style=" text-align: center " img src=" http://img1.17img.cn/17img/images/201805/noimg/0d88aade-7372-4d68-b5da-4d57efa64b73.jpg" title=" 001.jpg" width=" 650" height=" 478" border=" 0" hspace=" 0" vspace=" 0" style=" width: 650px height: 478px " / /p p style=" text-align: center " span style=" color: rgb(0, 112, 192) " strong 图:纳升级油-气-液滴(OAD)芯片和自动定位(SAM)装置的结构示意图(a)和用于单细胞蛋白质组分析的样品前处理和上样的全流程图(b)。 /strong /span /p p 蛋白质组学是后基因组计划之一,也是近年来的热点研究领域。作为生命体内功能直接执行者的蛋白质,和基因相比在揭示生命发育和疾病的发生发展的机制方面有更重大的意义。近年来,基于细胞群体内的蛋白质组学研究,因为不可避免地会将大量细胞内的信息平均化,已经越来越难以满足对生命功能更加深入探究的需要。从单细胞层面去了解细胞特征以及彼此之间的相互影响,可以为生物系统中细胞间的异质性提供更宝贵的信息,因此有着越来越大的迫切需求。 span style=" color: rgb(0, 112, 192) " 鉴于蛋白质不具有直接扩增的特性,以及蛋白质组学分析灵敏度的限制,目前对于少量乃至单细胞内蛋白质的检测在技术上还是极具挑战的。 /span /p p span style=" color: rgb(0, 112, 192) " 单细胞内蛋白质含量极少,就典型体细胞而言仅为0.1- 0.2 ng,远远小于常规蛋白质组学样品前处理所需要的微克级蛋白量。 /span 并且细胞内的蛋白质通常需要在离心管内完成复杂多步的前处理操作,包括细胞裂解和蛋白质释放、蛋白质沉淀纯化、蛋白质还原和烷基化、酶切等,一个全细胞裂解蛋白质组的最后反应体积均是几十甚至过百微升,在这个蛋白质组学常规前处理的过程中,由于样品与离心管的接触、多步的样品转移和不完全上样等原因,在进入质谱之前不可避免地带来了蛋白质的损失。用此流程来处理单细胞,即使之后结合液相色谱仪器的自动进样阀可以以小于十微升的体积完成上样也无济于事,因为尚未等到分离蛋白质样品就有可能已经损失大半了。 /p p style=" text-align: center " img src=" http://img1.17img.cn/17img/images/201805/noimg/ce546442-97dc-4a87-a243-558d62afe43e.jpg" title=" 003.png" / /p p span style=" color: rgb(0, 112, 192) " 如果说单细胞蛋白质组学是在米粒上雕刻,那么两个团队就是造出了适合在米粒上雕刻的工具刀。 /span span style=" color: rgb(0, 112, 192) " 该项研究起自2014年,两个团队的研究人员协同合作将微流控液滴技术与蛋白质组分析技术相结合,发展了一种微型化的油-气-液“三明治”芯片及相应的纳升级液体操控和进样方法,能够在原位静态的纳升级液滴中完成少量细胞蛋白质组学分析所必需的多步样品前处理操作,并且实现了将液滴样品直接高效地注入到色谱分析柱内完成后续的液相色谱分离与质谱检测。 /span 通过采用优化后的芯片材料、裂解试剂、酶切比例和色谱质谱参数等实验条件,该芯片系统可以分别成功地从100、50、10和1个HeLa细胞内鉴定到1360、612、192和51个蛋白质。更重要的是,研究人员首次实现了以单个鼠卵母细胞为初始样本的蛋白质组学分析,一共鉴定到355种蛋白质,其中存在一系列与生殖发育(Ovgp1和Pabpc1)、疾病相关(AnxA6,Kpna2,Cct6a和Pcbp2)的基因。在该工作中,还采用两种常规的基于离心管的前处理方法进行了对照实验,实验结果明确证实了微流控液滴体系具有更低的样品吸附损失、更高的酶切效率和更高效的疏水性蛋白质鉴定能力等明显优势,更适用于微量蛋白质样品的蛋白质组学分析。 /p p span style=" color: rgb(0, 112, 192) " 此项研究带来三个突破 /span :首先是成功发展了适合进行单细胞及类似微量样品的蛋白质组学分析样品前处理芯片和方法。芯片内550 纳升的液滴与芯片的接触面积仅为常规离心管模式下的十五分之一,显著减少了样品与反应器接触所带来的损失;二是发展了一种实现纳升级液滴的直接进样方法,利用3D打印加工的自动定位装置和高压气泵高效地(& gt 99%)将液滴样品注入到分析色谱柱内,完全避免了样品转移和经过液相仪器内复杂管路带来的损失;第三是为避免在常规微流控液滴系统中由于油相直接接触液滴而造成的缺陷,提出了一种采用气相来间隔液滴相和油相的新型液滴芯片结构,在成功防止液滴明显蒸发的同时,还避免了油相和液滴相直接接触带来的脂溶性样品的损失,也使系统能更方便地进行后续的分离柱进样、色谱分离和质谱检测。 /p p span style=" color: rgb(0, 112, 192) " 该项研究在基于质谱的单细胞及微量样品的蛋白质组学分析方面开启了全新的技术,其所具有的系统结构简单、容易搭建、操作方便、可靠性高等特点,使其有望被广泛应用到细胞间异质性的研究以及与临床相关的研究,包括稀有的循环肿瘤细胞分析、生殖细胞相关研究和疾病诊疗等方面,因此在未来具有巨大的持续开发和应用转化前景。 /span /p p style=" text-align: center " span style=" color: rgb(0, 112, 192) " img src=" http://img1.17img.cn/17img/images/201805/noimg/91e71d83-0fde-4b69-8b5f-10de6af33876.jpg" title=" 未命名_meitu_0.jpg" width=" 400" height=" 400" border=" 0" hspace=" 0" vspace=" 0" style=" width: 400px height: 400px " / /span /p p style=" text-align: center " strong span style=" color: rgb(0, 112, 192) " (左上:浙江大学 /span span style=" color: rgb(0, 112, 192) " 教授 /span span style=" color: rgb(0, 112, 192) " 方群、 /span /strong strong span style=" color: rgb(0, 112, 192) " 右上: /span span style=" color: rgb(0, 112, 192) " 北京大学 /span span style=" color: rgb(0, 112, 192) " 教授 /span span style=" color: rgb(0, 112, 192) " 黄超兰、 /span /strong strong span style=" color: rgb(0, 112, 192) " 左下: /span span style=" color: rgb(0, 112, 192) " 浙江大学博士生李紫艺、 /span span style=" color: rgb(0, 112, 192) " 右下:前国家蛋白质科学中心· 上海高级工程师 /span span style=" color: rgb(0, 112, 192) " 黄敏 /span span style=" color: rgb(0, 112, 192) " ) /span /strong /p p 该论文的第一作者为浙江大学博士生李紫艺,通讯作者为浙江大学化学系微分析系统研究所方群教授和北京大学医学部精准医疗多组学研究中心主任黄超兰教授。工作得到了国家自然科学基金和中国科学院杰出技术人才项目的支持。黄超兰教授的部分工作在前单位中科院国家蛋白质科学中心· 上海完成,特此鸣谢! /p p style=" line-height: 16px " img src=" /admincms/ueditor1/dialogs/attachment/fileTypeImages/icon_pdf.gif" / a href=" http://img1.17img.cn/17img/files/201805/ueattachment/b0d2802c-066e-4fdf-86c9-b683f1c317de.pdf" style=" color: rgb(0, 112, 192) text-decoration: underline " span style=" color: rgb(0, 112, 192) " Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis.pdf /span /a /p p span style=" color: rgb(0, 112, 192) " 文章链接: /span a href=" https://pubs.acs.org/doi/10.1021/acs.analchem.8b00661" _src=" https://pubs.acs.org/doi/10.1021/acs.analchem.8b00661" style=" color: rgb(0, 112, 192) text-decoration: underline " span style=" color: rgb(0, 112, 192) " https://pubs.acs.org/doi/10.1021/acs.analchem.8b00661 /span /a /p































 我要推广仪器
我要推广仪器
 下载APP
下载APP