对辐照食品的检测主要检测哪种物质的含量?使用什么方法测定?国家在这方面有强制性要求吗?
求助:气相色谱法主要检测蔗糖里面什么物质的含量?除了离子法检测二氧化硫,原子吸收检测重金属,请问还有什么?http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
感谢专家给我的答复。我想分别测试径流水样中的营养物质氮和磷的含量,主要是测定水样中的全氮和有效磷这两个指标,请问其测定方法主要有哪些?所使用的仪器设备是什么?
含量的高低会不会影响有关物质的高低,含量高,有关物质就会低,含量与有关物质之间的关系,有没有大佬解释一下,萌新化验员
在测量土壤项目时,需要土壤的干物质含量,有的项目标准中明确规定用新鲜土壤干物质含量,有的项目没有说明用哪一种土壤的干物质含量,有什么依据去判断是用新鲜土壤的干物质含量,还是用风干土壤的干物质含量,求各位大神帮忙解惑,万分感谢!!!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
在监测废水中总氮的含量时,前处理的方法啥样!主要就是有机物含量高的时候!!
要测釜残某物质的含量,它的含量很低,含着较大的水分,用气相怎么做呢?想知道它的含量
小弟最近在用NIR做一个低含量物质,物质质量百分含量在0.01-0.50%,在建模时总是发现线性不好(不是一条45度的直线,而几乎是一条0度的水平线),我知道对于NIR来说,低含量物质不太好检测,那么,1 对于特定的组分,我怎么确定它的检测下限呢?可以通过实验还是计算来获得么?2 我是觉得这是到了物质的检测下限,所以模型做不好,不知道各位专家有没有别的意见?3 另外,由于含量比较低,仪器的系统偏差是否也会影响到模型,那么,实验的时候仪器的偏差有多大,这个有办法测量或计算么?
麻烦各位,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检测钢铁某些物质含量?主要检的是什么?请详细说明!谢谢!
有关物质分析方法验证的可接受标准简介摘要:本文介绍了在对有关物质检查所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。 关键词:有关物质检查 分析方法验证 可接收标准 药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至一定的浓度范围内配制6份浓度不同的供试液,分别测定该杂质峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。 3.精密度 1)重复性 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于15%。 2)中间精密度 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于20%。 4.专属性 可接受的标准为:空白对照应无干扰,该杂质峰与其它峰应能完全分离,分离度不得小于2.0。 5.检测限 杂质峰与噪音峰信号的强度比应不得小于3。 6.定量限 杂质峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±5nm、流速相对值变化±20%以及采用三根不同批号的色谱柱进行测定时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:各杂质峰的拖尾因子不得大于2.0,杂质峰与其他成分峰必须达到基线分离;各条件下的杂质含量数据(n=6)的相对标准差应不大于2.0%,杂质含量的绝对值在±0.1%以内。 8、系统适应性 配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%,保留时间的相对标准差应不大于1.0%。另外,杂质峰的拖尾因子不得大于2.0,理论塔板数应符合质量标准的规定。 9.溶液稳定性 按照分析方法分别配置对照品溶液与供试品溶液,平行测定两次主成分与杂质的含量,然后将上述溶液分别贮存在室温与冰箱冷藏室(4℃)中,在1、2、3、5和7天时分别平行测定两次主成分与杂质的含量。 可接受的标准为:主成分的含量变化的绝对值应不大于2.0%,杂质含量的绝对值在±0.1%以内,并不得出现新的大于报告限度的杂质。含量测定分析方法验证的可接受标准简介摘要:本文介绍了在对含量测定所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。关键词:含量测定 分析方法验证 可接收标准在进行质量研究的过程中,一项重要的工作就是要对质量标准中所涉及到的分析方法进行方法学验证,以保证所用的分析方法确实能够用于在研药品的质量控制。为规范对各种分析方法的验证要求,我国已于2005年颁布了分析方法验证的指导原则。该指导原则对需要验证的分析方法及验证的具体指标做了比较详细的阐述。但是文中未涉及各具体指标在验证时的可接受标准,国际上已颁布的指导原则中也未发现相关的要求。另一方面,大多数药品研发单位在进行质量研究时,已逐步认识到分析方法验证的必要性与重要性,大都也在按照指导原则的要求进行分析方法验证,但验证完后却因没有一个明确的可接受标准,而难以判断该分析方法是否符合要求。本文结合国外一些大型药品研发企业在此方面的要求,提出了在对含量测定方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。1.准确度该指标主要是通过回收率来反映。验证时一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。2.线性线性一般通过线性回归方程的形式来表示。具体的验证方法为:在80%至120%的浓度范围内配制6份浓度不同的供试液,分别测定其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。3.精密度1)重复性配制6份相同浓度的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。2)中间精密度配制6份相同浓度的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于2.0%。4.专属性可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。5.检测限主峰与噪音峰信号的强度比应不得小于3。6.定量限主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。7.耐用性分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、流速相对值变化±20%时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。8、系统适应性配制6份相同浓度的供试品溶液进行分析,主峰峰面积的相对标准差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。本文为转帖!

今年我省土壤质量例行监测工作中的金属项目交给我们市站分析,我们结合水分含量折算了重金属含量。在分析含水率实验室时发现,干物质含量和水分含量之后略大于100%。 我当时感觉诧异,现试着解释这个看似不合理的实验现象。样品编号带盖容器重(g)容器+样品重(g)土壤水分含量土壤干物质含量水分含量+干物质含量烘干前烘干后135.68 45.63 45.43 2.05%97.99%100.04%230.63 40.75 40.59 1.61%98.42%100.03%329.80 40.42 40.24 1.72%98.31%100.03%428.43 38.68 38.49 1.89%98.15%100.04%531.53 41.53 41.48 0.50%99.50%100.00%628.78 38.81 38.73 0.80%99.20%100.01%726.90 37.13 37.04 0.89%99.12%100.01%827.69 38.48 38.43 0.47%99.54%100.00%930.69 40.76 40.71 0.50%99.50%100.00%1029.28 39.34 39.24 1.00%99.01%100.01%1130.72 40.74 40.55 1.93%98.10%100.04%1230.77 40.83 40.68 1.51%98.51%100.02%1330.19 40.67 40.24 4.28%95.90%[align=

磁性物含量测定1. 概述各种含铁矿物按其矿物组成,主要可分为四大类:磁铁矿、赤铁矿、褐铁矿和菱铁矿。磁铁矿是主要含铁矿物,其化学式为Fe3O4,其中FeO:31%, Fe2O3:69%。本方法采用磁选管法测定磁铁矿试样的磁性物含量。磁选管法的工作原理是在C行电磁铁的两极之间装有玻璃管,并作往复移动和旋摆运动。当磁选管中的试样通过磁场区时,磁性物即附着于管壁,非磁性物在机械运动中被水冲刷而排出,使磁性物与非磁性物分离。以磁性物和试样的百分比来表示磁性物含量。2. 试验主要设备:磁选仪(带磁选管),500ml烧杯,塑料桶,坩埚,烘箱,天平(精确到0.1mg),方形小磁铁等。http://ng1.17img.cn/bbsfiles/images/2013/07/201307271951_454093_1657564_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307271952_454095_1657564_3.jpg本实验的主要设备是磁选管。磁选管又名戴维斯管(Davis Tube)。它适用于选煤、矿山、冶金、地质等实验室。用来测定强磁性矿石的磁性成分含量。为矿石的分选提供参考数据。3. 操作步骤3.1 首先,检查电源是否正常,接线是否正确,水箱是否有水,玻璃管位置是否合适,手动盘车,确保设备运行正常。3.2 称取20g±20mg的样品,将试样装入一个容积为500mL的烧杯中,加入5~8mL的酒精和约400mL水,搅拌均匀,确保样品颗粒被充分地湿润。3.3 组装好全套装置后,接通电源,操作控制器,调节磁场至所需磁场强度值。磁场强度是根据磁性物磁性强弱及现场对磁性物要求来调节的。如果试样中磁性物很少或磁性物磁性较弱,则磁场强度应提高。一般将磁场强度设定在150~250mT之间。3.4 先用管夹夹紧玻璃管下端出口软管,向磁选管中加水直至距漏斗约5cm,以确保下一步骤所加磁性物悬浮于水中。3.5 将“电机启动开关”打开,此时,电机带动传动机构及玻璃管开始工作。然后将烧杯中的磁性物混合液体缓缓倒入漏斗,(玻璃管中液面不能太高,约距漏斗口处5cm,确保液体不从玻璃管上口溢出)同时打开玻璃管下部管夹,使液体缓缓流入塑料小桶中。3.6 待烧杯中磁性物混合液体全部倒入玻璃管后,再打开上面水箱的龙头,缓缓注入清水,确保磁性物悬浮于水中,而非磁性物质随水流下沉直至排出管外,磁性物颗粒在磁力作用下附着于管壁两磁极处,直至排出液体不含杂质。3.7 当排出液体不再含杂质时,停止加入清水,用管夹夹紧排水软管。将“电机启动开关”断开。电机停止工作。松开管夹。排出玻璃管内清水。3.8 断开“磁场启动开关”,当磁场显示为“0000”后,将玻璃管拆下,在玻璃管出口处放一个干净的500ml烧杯,轻轻转动玻璃管,同时用洗瓶从玻璃管上口冲刷,把磁性物从玻璃管中冲洗干净,收集到烧杯中。3.9 将装有磁性物混合液的烧杯静置约15分钟,直至磁性物沉淀,上部水澄清,慢慢倒出烧杯中的水,同时用一块强磁铁放在烧杯底部,以防止杯中磁性物有任何损失。3.10 开激磁电源,关闭螺旋夹,向磁选管中加水,打开螺旋夹,并使水流动,把第一个塑料小桶中的液体和固体慢慢加入漏斗,并使混合液通过磁选管进入第二个塑料小桶中。将第二次收集到的磁性物质和第一次的合并在一起。将磁性物质转入干燥的并已称好重量为M0的碗型坩埚中。注:
请问物质的含量和物质的纯度是同一个概念吗?有的物质写含量达到某值,又写一个含量,到底是什么意思呢?谢谢各位:)[em01]
专家您好!“某物质的含量是100%”我所配制的染料:要测的那种物质含量肯定在2%-4%之间,所以我还是想请教专家!谢谢
求助:如何在线检测粉浆干物质含量测量介质是酒精发酵来料:小麦粉和玉米、木薯粉混合粉浆,要求控制来料干物质含量。目前采用的是化验室取样,物理干燥法,烘干水分测得干物质含量(质量百分比)。粉浆是物料加水常温混合,实际应是悬浊液,取样放置一会儿即固液分离沉淀了。我想请教对这种介质有没有在线检测或现场快速检测干物质含量的仪器或方法呢,谢谢!
常温下为固体的物质,在检测时加入一种溶剂与之混溶,但是有一个问题,就是加入溶剂之后如何对主要组份进行计算(我们对含量的要求在ppm级)谢谢各位专家的帮忙。
各位兄弟姐妹: 大家平时做有关物质、含量样品检测时时怎么操作的?我们 含量测定样品称取各两份,一份进两针,一份进三针,然后五针计算RSD 有关物质只称取一份,进五针。 觉得挺麻烦的。大家都是怎么弄的?
不知道哪位可以帮忙,给点意见?我最近接到一个实验,主要是做锡炉中锡膏含量的变化。Sn96.5% Ag3% Cu0.5%的锡膏,可能还含有Cd,Pb微量,随着锡膏的使用,一段时间后,锡膏中锡含量会有所变化。关键问题是:客户要求做锡含量测定,但锡含量过高,我觉得不好测。而先测定Ag,Cu含量及Pb,Cd,用总含量来减去以上元素含量,估算出锡的含量,我又觉得不够专业也不够有把握。请帮忙想想合适的测试方法。谢谢!
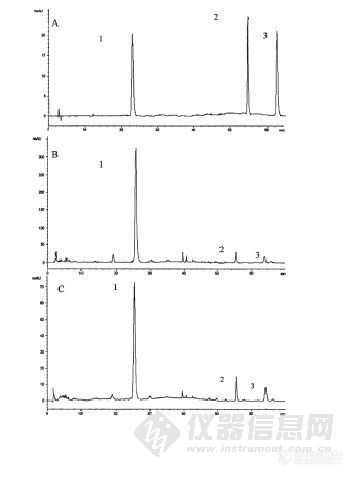
作者:王洋(南京中医药大学)摘要:本文对不同采收期、不同贮存时间的广陈皮药材主要成分的含量进行了测定,归纳总结了其主要的变化规律,旨在揭示个青皮与广陈皮药材“同源不同性”的机理以及初步探索贮存时间对广陈皮内在成分的影响,为广陈皮药材的临床应用提供实验依据。 1.本文所需广陈皮药材样品共13份,收集于广东省新会县广陈皮种植基地,样品分别为2004年11月、2005年10月、2005年11月、2005年12月、2006年10月、2006年11月、2006年12月、2007年5/6月、2007年8月、2007年9月、2007年10月、2007年11月和2007年12月。 2.对各广陈皮样品的主要成分进行了含量测定,确定了测定方法。总黄酮、总生物碱和总多糖采用紫外分光光度法,回归方程、相关系数和线性范围分别为:y=3.397x+0.011,r=0.9998,线性范围:0.04mg/m1~0.24mg/ml;y=7.12x+0.018,r=0.9998,线性范围:0~0.18mg/ml;y=0.083x-0.005,r=0.9997。挥发油成分采用GC/MS法测定,气相色谱条件:色谱柱为DB-5(30m×250μm×0.25μm)石英毛细管色谱柱;进样口温度220℃;程序升温60℃(维持5min),以5℃/min升温至200℃;载气为高纯氦气,流量1mL/min,溶剂延迟3min。质谱条件:MSD离子源为El源,离子源温度230℃,电子能量70eV,扫描质量范围50~550质量数。加速电压1000eV。 橙皮苷、川陈皮素和橘皮素的含量测定采用HPLC法,色谱条件为:流动相体系为甲醇(A)-乙腈(B)-4%醋酸水溶液(C),梯度洗脱。0~25min,A: B:C=10:15:75;25~40nun,A升至50%,A:B:C=50:15:35,保持30min。色谱柱为Platisil ODS柱(5μm,250×4.6mm),柱温30℃,流速1ml/min。检测波长:0~50min,283nm;50~70min,332nm。 3.对不同采收期广陈皮药材主要成分的含量变化进行了对比,结果为:个青皮总黄酮和橙皮苷的含量最高,而广陈皮药材挥发油、生物碱以及多糖类成分均高于个青皮,这可能就是个青皮与广陈皮“同源不同性”的内在机理。 对于不同贮存时间的广陈皮药材,黄酮类成分与总生物碱含量随着贮存时间的延长会基本保持不变或者略有上升。 4.用“老化”模拟实验处理广陈皮药材,结果表明在高温条件下,黄酮类成分化学性质较稳定,且随着烘烤时间的延长,含量会略有上升。另外,在高温条件下,药材内部可能发生了化学成分的转化并产生新的极性较大的物质。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131431_383497_1606903_3.jpg
用标准加入法测定待测物质含量需要跟外标法一样得到一个方程吗?如果也是得到一个方程,这个方程就可以用于后面精密度、加样回收率的验证了?
最近在帮公司测一个药物新剂型的含量,按照公司的任务标准,除了要测主药物含量外,还要测有关物质的。任务书上是这样写的,测1%和1000%的有关物质,请问这是什么意思呢?对于测有关物质有没有统一的标准,比如说要测多大的浓度?分析多长时间?
外标法测量物质的含量时标液的配置问题:1.是每天现配还是配置好用一段时间,用一段时间的标准怎么判断,方法学验证吗。2.每次配几个标液。3.有没有相关指导文件。
[color=#444444]我想用液相色谱方法测定一个混合物中主物质和杂质对氯苯胺的含量,目前只能买到对氯苯胺这个标样,主物质是没法买到标样的,试问除了归一化法,还有什么方法可以测定主物质的含量呢?可否有从对氯苯胺入手的方法?谢谢各种前辈的建议![/color]
如果制剂的含量测定和有关物质检查的条件一样,在做方法学比如线性,准确度(加入回收率)的时候,可不可以一起做,还是先做有关物质,然后做含量测定?
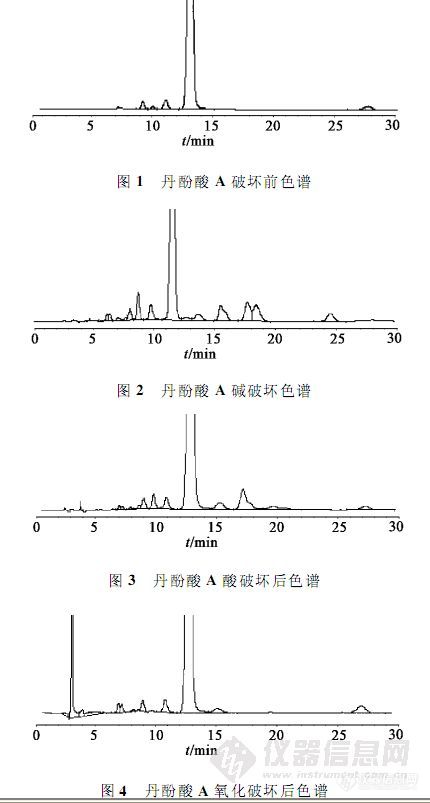
【作者】 于翠翠; 刘军锋; 车鑫; 刘珂;【机构】 烟台大学药学院; 山东靶点药物研究有限公司;【摘要】 目的:建立一种测定丹酚酸A中有关物质含量的方法。方法:用外标法测定丹酚酸C的含量,采用Diamonsil C18柱(4.6 mm×250 mm,5μm),乙腈-0.2%磷酸水溶液(30∶70)为流动相,流速1 mL.min-1,检测波长285 nm,柱温30℃,以主成分自身对照法控制其他杂质的总含量。结果:丹酚酸C的含量均1.0%,其他杂质峰面积和3.0%。结论:该方法专属性强,灵敏度高,简单准确,能有效控制丹酚酸A中有关物质的含量。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207231559_379228_2379123_3.jpg
师哥做的实验,测生物质厌氧发酵挥发性有机酸含量。采用GC-2014C[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url](日本岛津)分析,FID检测器,WondaCap FFAP色谱柱,梯度升温,载气为N2。这一批实验结果为何只有总酸含量,却没有具体酸含量。
做影响因素试验哦 有关物质有一定的变化规律 可是做出来的含量却是高高低低 不能与之保持相对一致 很困惑 这样的含量做出来还有意义么







 我要推广仪器
我要推广仪器
 下载APP
下载APP