石油的馏分组成 由于石油是由具不同沸点的烃化合物混合而成,因此通过控制不同的温度而可分别获得不同的石油产品[color=#333333][font=&][color=#333333]石油是一个多组分的复杂混合物,其沸点范围很宽,从常温一直到500℃以上。所以,无论是对石油进行研究或进行加工利用,都必须对石油进行分馏。分馏就是按照组分沸点的差别将石油“切割”成若干“馏分”,例如500℃的油称为减压渣油(简称VR); 同时人们也将常压蒸馏后大于350℃的油称为常压渣油或常压重油(简称AR)。下表是国内外部分原油的馏分组成。国内外部分原油的馏分组成[/color][table][tr][td=1,2]原油名称[/td][td=4,1]馏分组成(质量分数),%[/td][/tr][tr][td=1,1,135]初馏点~200℃[/td][td=1,1,135]200℃~350℃[/td][td=1,1,135]350℃~500℃[/td][td=1,1,135]500℃[/td][/tr][tr][td=1,1,135]大庆[/td][td=1,1,135]11.5[/td][td=1,1,135]19.7[/td][td=1,1,135]26.0[/td][td=1,1,135]42.8[/td][/tr][tr][td=1,1,135]胜利[/td][td=1,1,135]7.6[/td][td=1,1,135]17.5[/td][td=1,1,135]27.5[/td][td=1,1,135]47.4[/td][/tr][tr][td=1,1,135]孤岛[/td][td=1,1,135]6.1[/td][td=1,1,135]14.9[/td][td=1,1,135]27.2[/td][td=1,1,135]51.8[/td][/tr][tr][td=1,1,135]辽河[/td][td=1,1,135]9.4[/td][td=1,1,135]21.5[/td][td=1,1,135]29.2[/td][td=1,1,135]39.9[/td][/tr][tr][td=1,1,135]华北[/td][td=1,1,135]6.1[/td][td=1,1,135]19.9[/td][td=1,1,135]34.9[/td][td=1,1,135]39.1[/td][/tr][tr][td=1,1,135]中原[/td][td=1,1,135]19.4[/td][td=1,1,135]25.1[/td][td=1,1,135]23.2[/td][td=1,1,135]32.3[/td][/tr][tr][td=1,1,135]新疆(输油管)[/td][td=1,1,135]15.4[/td][td=1,1,135]26.0[/td][td=1,1,135]29.9[/td][td=1,1,135]29.7[/td][/tr][tr][td=1,1,135]新疆(库尔勒)[/td][td=1,1,135]19.6[/td][td=1,1,135]31.1[/td][td=1,1,135]26.1[/td][td=1,1,135]23.2[/td][/tr][tr][td=1,1,135]新疆(九区)[/td][td=1,1,135]2.3[/td][td=1,1,135]18.9[/td][td=1,1,135]28.9[/td][td=1,1,135]49.9[/td][/tr][tr][td=1,1,135]单家寺[/td][td=1,1,135]1.2[/td][td=1,1,135]12.2[/td][td=1,1,135]18.3[/td][td=1,1,135]68.3[/td][/tr][tr][td=1,1,135]沙特(轻质)[/td][td=1,1,135]23.3[/td][td=1,1,135]26.3[/td][td=1,1,135]25.1[/td][td=1,1,135]25.3[/td][/tr][tr][td=1,1,135]沙特(轻重混合)[/td][td=1,1,135]20.7[/td][td=1,1,135]24.5[/td][td=1,1,135]23.2[/td][td=1,1,135]31.6[/td][/tr][tr][td=1,1,135]阿联酋(麦瑞波)[/td][td=1,1,135]31.5[/td][td=1,1,135]30.6[/td][td=1,1,135]23.2[/td][td=1,1,135]14.7[/td][/tr][tr][td=1,1,135]英国(北海)[/td][td=1,1,135]29.0[/td][td=1,1,135]27.6[/td][td=1,1,135]25.4[/td][td=1,1,135]18.0[/td][/tr][tr][td=1,1,135]印尼(米纳斯)[/td][td=1,1,135]11.9[/td][td=1,1,135]30.2[/td][td=1,1,135]24.8[/td][td=1,1,135]33.1[/td][/tr][/table]与国外原油相比,我国主要油区原油中的大于500℃减压渣油的含量较高,小于200℃的汽油馏分含量较少。原油中的汽油馏分含量低、渣油含量高是我国原油馏分组成的一个特点。从石油直接分馏得到的馏分称为直馏馏分,它们基本上保留着石油原来的性质,例如基本上不含不饱和烃。石油直馏馏分经过二次加工(如催化裂化等)后,所得的馏分与相应直馏馏分的化学组成不同,例如催化裂化产物的化学组成中就含有不饱和烃(并非一切二次加工产物都含有不饱和烃)。
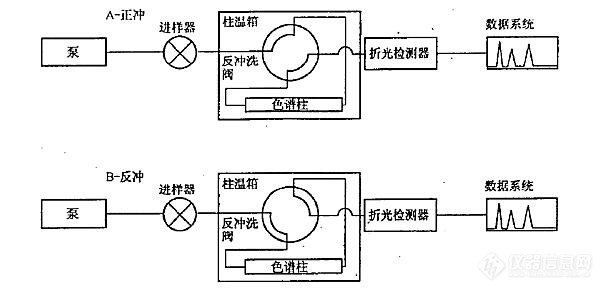
[b][font=宋体]问题描述:使用[/font]SH 0806-2008[font=宋体]标准方法测定柴油和馏程范围为[/font]150~400[font=宋体]℃[/font][font=宋体]的石油馏分中单环芳烃、双环芳烃、三环以上芳烃和多环芳烃含量,有哪些注意事项?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])系统结构原理如图[/font]6-41[font=宋体]所示,系统使用正庚烷做流动相,单输送泵、极性色谱柱(氨基或者氰基柱)、四通阀和示差检测器实现分离[/font].[align=center][img=,605,292]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047269515_6030_3389662_3.jpg!w605x292.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-41 SH 0806 [font=宋体]方法的硬件原理[/font][/i][/align][font=宋体]([/font]2[font=宋体])待机状态和进样状态下(即状态[/font]A[font=宋体]),流动相自右向左流过极性色谱柱,柴油中的烃类和芳烃类组分实现分离。理想情况下,单环芳烃、双环芳烃、多环芳烃依次在色谱柱出口流出,如图[/font]6-42[font=宋体]所示。[/font][align=center][img=,508,274]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047383890_9606_3389662_3.jpg!w508x274.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-42 [font=宋体]理想的色谱柱内样品分离谱图[/font][/i][/align][font=宋体]([/font]3[font=宋体])当双环芳烃流出色谱柱后(色谱图中[/font]A[font=宋体]点位置,或称为切换点),四通阀旋转,系统状态变为反吹。色谱柱内流动相的方向变成自左至右,将三环以及三环以上的芳烃类物质反吹出色谱柱,在示差检测器上表现为单峰,如图[/font]6-43[font=宋体]所示。[/font][align=center][img=,536,271]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047483421_9431_3389662_3.jpg!w536x271.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-43[font=宋体]最终样品分离谱图[/font][/i][/align][font=宋体]([/font]4[font=宋体])该方法的原理是比较理想化的,干扰因素也比较多。柴油中的单环芳烃、双环芳烃、多环芳烃是否可以清晰彻底的分离开,是难以保证的。况且柴油中的二烯烃、杂环类、酯类化合物等都会对分析结果带来影响。[/font][font=宋体]([/font]4[font=宋体])色谱柱的选择十分重要,具体的选型需要咨询色谱柱厂家。[/font][font=宋体]([/font]5[font=宋体])切换点的选择非常重要。样品组成可能比较复杂,[/font]A[font=宋体]点可以选择的时间窗口就会比较窄,需要多次重复实验寻找合适的切换点。分析条件需要非常稳定,需要较为严格的控制流动相组成、泵输送流速以及色谱柱温度,以免影响保留的重复性。[/font][font=宋体]([/font]6[font=宋体])需要注意流动相不稳定导致保留时间漂移:[/font][font=宋体]保留时间的漂移是最为常见的问题。在进样系统性能测试标准样品时,芳烃的保留时间长时间的漂移,致使难以确定切换点。往往会耗费较多时间来平衡系统,等待保留时间稳定,从而降低分析效率。其本质的原因在于流动相的不稳定。[/font]SH 0806-2008[font=宋体]系统分离原理属于正相液相色谱,我们知道正相[/font]HPLC[font=宋体]一般不太容易得到良好的保留时间重复性。原因是在正相[/font]HPLC[font=宋体]分析中,流动相中的微量水会显著的改变其极性。假设流动相原先的极性为[/font]0.01[font=宋体],吸收微量水之后极性变为[/font]0.02[font=宋体],看上去似乎变化不大,但其实极性增大了一倍。尤其是使用硅胶色谱柱的场合,流动相与环境空气中的水蒸气发生交换,改变了极性,从而影响保留时间。在使用硅胶柱分析时,一般要避免使用彻底干燥的正己烷流动相,避免吸水造成保留不稳定,甚至需要特意在流动相中加入微量的水。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font][color=red] [/color]
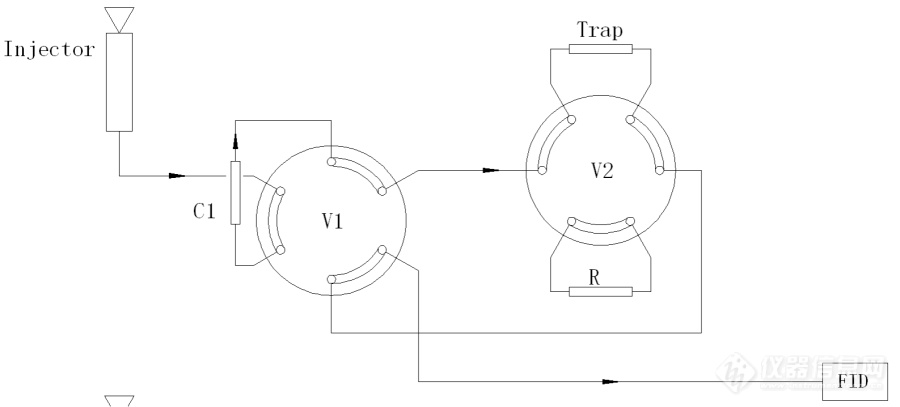
[align=center][size=24px]GBT 30519 轻质石油馏分和产品中烃族组成和苯的测定-多维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的原理解析[/size][/align][align=center][/align][align=center][/align][align=center][color=black]概述[/color][/align][align=center][color=black][/color][/align][align=center][color=black]《GBT 30519 轻质石油馏分和产品中烃族组成和苯的测定-多维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[/color][/align][color=black]》分析原理图解。[/color][align=center][color=black]一 背景介绍[/color][/align][color=black]溶剂油、汽油产品、汽油调和组分等样品中均含有一定量的烯烃、芳烃类物质。以车用汽油为例,这些物质是提高辛烷值的重要添加物。但是芳烃和烯烃的含量过低或者过高会均造成环境污染、影响发动机性能等问题,所以需要对样品中的总烯烃类、总芳烃和苯的含量有一定的限制。GBT30519方法即用以测定此类样品中饱和烃类、烯烃类、芳烃类组分含量。[/color][align=center][color=black]二 结构原理[/color][/align][color=black]GB30519多维[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析系统的结构如图1所示,系统由两个自动六通阀V1、V2和强极性预分离色谱柱C1、烯烃捕集柱Trap、平衡阻尼柱R组成。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]系统分析程序对两个工作于不同温度下的六通阀进行精确、定时的切换,改变系统中三根色谱柱的反吹和连接状态,将样品中的饱和烃类、烯烃类、芳烃类(C7以上芳烃)和苯各类烃族组成予以测定。[/color][color=black]系统采用校正面积归一法进行测定。汽油的醇醚类物质会对分析结果带来一定的影响,醇类物质会与C7以上的芳烃类同时出峰,醚类物质会与烯烃类无疑同时出峰,所以该系统测定普通车用汽油时,需要与SH 0663汽油中氧化物测定系统协同工作。需要预先获知样品中的醇醚类物质含量,以校正族组成结果。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941173333_9697_1604036_3.jpg[/img][/align][align=center]图1 GBT30519 系统硬件结构(系统待机状态)[/align][align=center][color=black]三 工作流程[/color][/align][color=black]该系统的工作流程如下:[/color][color=black]进样,饱和烃出峰:[/color][color=black]在图1所示的系统状态下,0.1ul的汽油样品直接进样至[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的进样口(Injector)中,样品气化并进入预切色谱柱(C1)和烃类捕集阱(Trap),系统的简化结构如图2所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941176799_6958_1604036_3.jpg[/img][/align][align=center]图2 进样状态下系统结构简化示意图[/align][color=black]汽油样品中各类烃族组分在预切色谱柱(Column-1)内的分布状态,如图3所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941180265_1689_1604036_3.jpg[/img][/align][align=center]图3 预切柱流出组分分布状态[/align][color=black]在强极性预切色谱柱(一般为高含量的BCEF固定相的填充柱)中汽油中各组分被分离成为大致三组:轻烃类组分——包括样品中的饱和烃和烯烃类物质、苯和重芳烃类物质。[/color][color=black]饱和烃类和烯烃类组分流经烯烃捕集阱(Trap)时,烯烃类物质被吸附在Trap中,只有饱和烃类物质流出色谱柱,此时在FID检测器可以观察到饱和烃类的色谱峰。[/color][color=black]第一次切换,苯出峰[/color][color=black]当饱和烃类物质全部流出色谱柱C1后,色谱系统控制六通阀V2旋转,系统状态发生变化,如图4所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941183557_9929_1604036_3.jpg[/img][/align][align=center][color=black]图4 系统第一次切换状态[/color][/align]此时,烯烃捕集柱被封闭起来。系统的简化结构如图5所示:[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941184729_2755_1604036_3.jpg[/img][/align][align=center]图5 第一次切换状态的系统的简化结构[/align][color=black]预切色谱柱(C1)中的苯,经过阻尼柱R之后流出进入FID检测器出峰。[/color][color=black]第二次切换,重芳烃出峰[/color][color=black]当苯全部流出色谱柱C1后,色谱系统控制六通阀V1旋转,系统状态发生变化,如图6所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941183478_7214_1604036_3.jpg[/img][/align][align=center][color=black]图6 系统第二次切换状态[/color][/align]此时系统的简化结构如图7所示:[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941186741_3952_1604036_3.jpg[/img][/align][align=center]图7 第二次切换状态的系统的简化结构[/align][color=black]系统第二次切换之后,预切色谱柱(C1)载气流向发生反转,色谱柱内的重烃类被反吹经过阻尼柱R之后流出进入FID检测器,在最终谱图上表现为一个色谱峰(这个色谱峰未必形状比较规整)。[/color][color=black] 第三次切换,烯烃类出峰。[/color][color=black]当重烃类组分全部反吹流出色谱柱C1后,色谱系统控制六通阀V2旋转,系统状态发生变化,如图8所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941185373_7173_1604036_3.jpg[/img][/align][align=center][color=black]图8 系统第三次切换状态[/color][/align]此时系统的简化结构如图9所示:[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941186467_2507_1604036_3.jpg[/img][/align][align=center]图9 第三次切换状态的系统的简化结构[/align][color=black]系统第三次切换之后,烯烃捕集阱(Trap)载气流向发生反转,色谱柱内捕集的烯烃类被反吹进入FID检测器,在最终谱图上表现为一个色谱峰。系统谱图如图10所示:[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110010941189485_9302_1604036_3.jpg[/img][/align][align=center]图10 系统谱图[/align]系统复位,等待下次进样。
[font=Encryption][color=#898989]摘要:[/color][/font][font=Encryption][color=#666666] 哈萨克斯坦(哈国)原油是中国西北口岸进口的主要原油,实沸点切取350~400,400 ~ 450和450~520℃三个馏分按润滑油指标进行性质分析,表明哈国原油润滑油馏分黏度指数较高,链状烃含量在65%左右,芳烃含量小,黏度指数高,溶剂精制难度不大,精制油收率高,是比较理想的润滑油基础油原料.把350~520℃馏分按催化裂解原料进行分析,表明哈国原油减压馏分平均分子中烷基侧链上的碳原子分数为66.17%.重金属镍加钒含量少(0.16μg,/g),可直接作为重油催化裂化的原料.但该馏分硫含量高,催化裂化汽油、柴油应加氢精制.在加压微反色谱装置上对哈国原油350~ 520℃馏分进行催化裂化实验,结果表明该馏分转化率高达70.27%,轻质油收率高达64.51%,总液收率高达80.74%,表明哈国原油350~520℃馏分是优质催化裂化原料.[/color][/font]
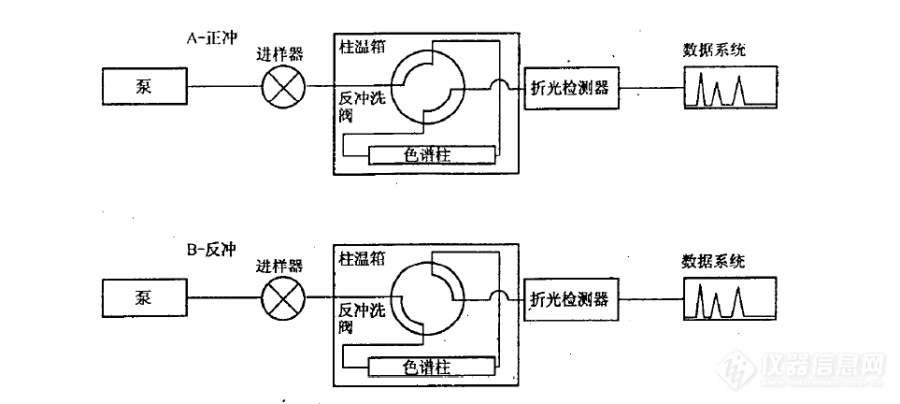
[align=center][font=宋体][size=14.0000pt]SH 0806-2008 [font=宋体]中间馏分芳烃含量的测定 分析方法的注意事项[/font][font=Calibri]----[/font][font=宋体]流动相[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]概述:[/font]SH0806-2008[font=宋体]标准使用高效液相色谱方法测定柴油和馏程范围为[/font][font=Calibri]150-400[/font][font=宋体]℃的石油馏分中单环芳烃、双环芳烃、三环以上芳烃和多环芳烃含量。该分析方法虽然比较简单,但是分析条件的控制比较重要。[/font][/size][/font][font=宋体][size=12.0000pt]在实验操作中,尤其需要注意流动相的问题,流动相不良会造成保留时间漂移从而使得色谱柱切换时间选择发生困难。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]一[/font] [font=宋体]原理介绍[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]系统结构原理如图[/font]1[font=宋体]所示,系统使用正庚烷做流动相,单输送泵、极性色谱柱(氨基或者氰基柱)、四通阀和示差检测器实现分离。[/font][/size][/font][font=宋体][size=12.0000pt][font=宋体]待机状态和进样状态下(即状态[/font]A[font=宋体]),流动相自右向左流过极性色谱柱,柴油中的烃类和芳烃类组分实现分离。理想情况下,单环芳烃、双环芳烃、多环芳烃依次在色谱柱出口流出,如图[/font][font=Calibri]2[/font][font=宋体]所示。[/font][/size][/font][font=Calibri][size=12.0000pt][img=,690,309]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010841562718_6781_1604036_3.png!w690x309.jpg[/img] [/size][/font][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]1 SH 0806 [font=宋体]方法的硬件原理图[/font][/size][/font][/align][align=center][font=Calibri][size=12.0000pt][img=,690,348]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010842112720_7230_1604036_3.png!w690x348.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]2 [font=宋体]色谱柱内样品理想分离示意图[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]当双环芳烃流出色谱柱后(色谱图中[/font]A[font=宋体]点位置,或称为切换点),四通阀旋转,系统状态变为反吹。色谱柱内流动相的方向变成自左至右,将三环以及三环以上的芳烃类物质反吹出色谱柱,在示差检测器上表现为单峰,如图[/font][font=Calibri]3[/font][font=宋体]所示。[/font][/size][/font][align=center][font=Calibri][size=12.0000pt][img=,690,351]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010842234040_3244_1604036_3.png!w690x351.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]3 [font=宋体]最终谱图[/font][/size][/font][/align][align=center][font=宋体][size=12.0000pt][font=宋体]二[/font] [font=宋体]分析注意要点[/font][/size][/font][/align][font=宋体][size=12.0000pt]首先要注意该方法的原理是比较理想化的,干扰因素也比较多。柴油中的单环芳烃、双环芳烃、多环芳烃是否可以清晰彻底的分离开,是难以保证的。况且柴油中的二烯烃、杂环类、酯类化合物等都会对分析结果带来影响。[/size][/font][font=宋体][size=12.0000pt]其次,色谱柱的选择十分重要,具体的选型需要咨询色谱柱厂家。[/size][/font][font=宋体][size=12.0000pt]再次,切换点的选择非常重要。[/size][/font][font=宋体][size=12.0000pt][font=宋体]样品组成可能比较复杂,[/font]A[font=宋体]点可以选择的时间窗口就会比较窄,需要多次重复实验寻找合适的切换点。[/font][/size][/font][font=宋体][size=12.0000pt]分析条件需要非常稳定,需要较为严格的控制流动相组成、泵输送流速以及色谱柱温度,以免影响保留的重复性。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]三[/font] [font=宋体]常见问题[/font]——流动相[/size][/font][/align][font=宋体][size=12.0000pt]保留时间的漂移是最为常见的问题。在进样系统性能测试标准样品时,芳烃的保留时间长时间的漂移,致使难以确定切换点。往往会耗费较多时间来平衡系统,等待保留时间稳定,从而降低分析效率。[/size][/font][font=宋体][size=12.0000pt]其本质的原因在于流动相的不稳定。[/size][/font][font=宋体][size=12.0000pt][font=宋体]笔者曾经在使用该系统时,长时间重新系统后,连续进样[/font]10[font=宋体]余次系统测试标样,发现芳烃的保留时间不断发生缩短,认为色谱柱未彻底平衡。[/font][/size][/font][font=宋体][size=12.0000pt][font=宋体]第二天更换了流动相(新换的流动相没有彻底封口放置在实验室[/font]10[font=宋体]小时),进样[/font][font=Calibri]3[/font][font=宋体]次,芳烃的保留时间较为稳定。[/font][/size][/font][font=宋体][size=12.0000pt]后来考虑了一下,原因应当为新换的正庚烷中水含量已经与空气中的水含量交换平衡,或者说水含量已经比较稳定,进而使得芳烃保留稳定。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]四[/font] [font=宋体]小结[/font][/size][/font][/align][font=宋体][size=12.0000pt]SH 0806-2008[font=宋体]系统分离原理属于正相液相色谱,我们知道正相[/font][font=Calibri]HPLC[/font][font=宋体]一般不太容易得到良好的保留时间重复性。原因是在正相[/font][font=Calibri]HPLC[/font][font=宋体]分析中,流动相中的微量水会显著的改变其极性。假设流动相原先的极性为[/font][font=Calibri]0.01[/font][font=宋体],吸收微量水之后极性变为[/font][font=Calibri]0.02[/font][font=宋体],看上去似乎变化不大,但其实极性增大了一倍。[/font][/size][/font][font=宋体][size=12.0000pt]尤其是使用硅胶色谱柱的场合,流动相与环境空气中的水蒸气发生交换,改变了极性,从而影响保留时间。在使用硅胶柱分析时,一般要避免使用彻底干燥的正己烷流动相,避免吸水造成保留不稳定,甚至需要特意在流动相中加入微量的水。[/size][/font]

过硅胶柱层析收集了96个馏分,结果没有一个馏分检测到所需的效果的!想到以下原因:1、上柱前的样品是冷冻干燥后的粉末,可能这些物质在冷冻过程中丧失了活性;2、收集的馏分过分稀释了目标物质;3、在检测馏分的活性前,需要将馏分中的洗脱剂(氯仿和甲醇)旋转蒸发除去,然后用去离子水洗净蒸馏瓶的内壁,可能在此过程中损失较大;4、最坏的可能,目标物根本没洗下来。1和2两个原因已经排除,求助各位高手,要避免3这个可能,可以怎样改进除去洗脱剂的方法,保证物质不会损失?至于第4个原因,我上传几张洗脱前后的硅胶柱照片,各位高手帮忙判断下其中的物质是否洗下来了?http://ng1.17img.cn/bbsfiles/images/2011/02/201102212210_278729_2220018_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/02/201102212213_278731_2220018_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/02/201102212214_278732_2220018_3.jpg是否还有其它原因?急望高手指导。
谁用过岛津的馏分自动馏分收集器 如何确定收集馏分时间 和延迟时间 还有怎么判断馏分是否收集完整 我现在一直都是出锋前一分钟和出锋后一分钟 一共三分钟 感觉收集的不完全 做出样品比原来的低好多 只能加标测试回收率看吗
[color=#013add][size=5]制备型液相色谱有馏分收集器,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]也有制备馏分收集器,你用过哪些呢?[/size][/color]
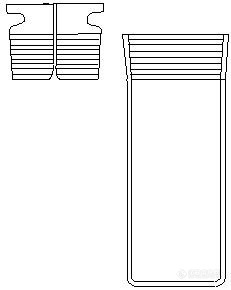
李军芳、杜淑凤/煤焦油及馏分油的密度是煤焦油评价非常重要的基础数据之一。测定煤焦油馏分油密度可近似地评价其质量和化学组成。从化学组成看,烷烃的密度最小,环烷烃居中,芳烃的密度大,含胶质和沥青质多的油品密度更大。目前测定油品密度的方法通常有密度测定仪法、比重瓶法和密度计法。对常温下为液态的不含固煤焦油轻质馏分油,采用数字手持式密度计测量,该方法方便、快捷、精确。针对常温下为凝固态或固态的煤焦油及重质馏分油,采用广口型比重瓶(图1)进行密度的分析测定。对于测量温度下为非凝固态的煤焦油,可采用密度计法,按照GB/T 2281-2008的标准方法测量。[align=center][img=,231,290]http://ng1.17img.cn/bbsfiles/images/2017/08/201708281514_01_3232859_3.jpg[/img][/align][align=center]图1 广口型比重瓶[/align]下面介绍下广口型比重瓶的操作步骤:1. 比重瓶20 ℃水值的测定1.1 将仔细洗涤,干燥并冷至室温的比重瓶称量精确至0.0001g,空比重瓶质量记为m1。1.2 用注射器将新煮沸并冷却至18~20 ℃的蒸馏水装满至比重瓶顶端,加上塞子,然后放入20℃的恒温水浴中,至少保持30 min,但不要浸没比重瓶或毛细管上端。待温度达到平衡,没有气泡,液面不再变动时,取出比重瓶,用一块清洁的无毛布擦干比重瓶的外壁,并将毛细管顶部过剩的水轻轻擦去,消除静电后称量精确至0.0001 g,装有水的比重瓶质量记为m2。1.3 比重瓶的20 ℃水值m20按式(1)计算:[align=center]m20=m2-m1 …………………………… (1)[/align]式中: m20 —— 比重瓶20 ℃的水值,g; m2—— 装有20 ℃水的比重瓶质量,g; m1—— 空比重瓶质量,g。 比重瓶的水值应测定3~5次取其算术平均值作为该比重瓶的水值。2. 样品的密度测定2.1 将已知水值的比重瓶称量精确至0.0001 g,空比重瓶质量记为m1。2.2 对煤焦油及煤焦油重质馏分试样,最好采用加入半瓶试样,勿使瓶壁污浊。如试样为脆性固体(如沥青),则粉碎或熔化后装入,然后用加热,抽空等办法以除去气泡,冷却到接近20 ℃。将上述比重瓶称量精确至0.0001 g,得到装有半瓶试样的比重瓶质量m3。2.3 用蒸馏水充满上述比重瓶。并放在20 ℃的恒温水浴中,恒温时间不少于30 min,待温度达到平衡,没有气泡,液面不再变动时,取出比重瓶,用一块清洁的无毛布擦干比重瓶的外壁,并将毛细管顶部过剩的水轻轻擦去,消除静电后称量精确至0.0001 g,得到装有半瓶试样和水的比重瓶质量m4。2.4 样品密度ρ20结果计算[align=center][img=,281,45]http://ng1.17img.cn/bbsfiles/images/2017/08/201708281518_01_3232859_3.jpg[/img]…………(2)[/align]式中: m3—在20℃时装有半瓶试样的比重瓶质量,g; m1—空比重瓶质量,g; m20—在20℃时比重瓶的水值,g; m4—在20℃时装有半瓶试样和水的比重瓶质量,g; 0.99820 —水在20 ℃的密度,g/cm3; 0.0012— 在20 ℃、大气压为760毫米汞柱时空气的密度,g/cm3。
我第一次用硅胶柱层析,洗脱剂为:氯仿、甲系统,收集的洗脱馏分蒸发浓缩后,能在薄层板上检测到物质,但是由于上样量太多,后来的洗脱梯度没用。第二次分离的洗脱洗脱与第一次相同,但收集的馏分蒸干后用PBS缓冲液洗净,结果却在硅胶板上什么都检测不到。我以为原因是点板的溶剂不同(一个是氯仿甲醇洗脱剂,一个是PBS缓冲液),所以用其中一个馏分做了两种溶剂的薄层层析,结果还是什么都没有。求助各位高手,这是什么原因?如果在板上没有检测到物质,是否就说明该馏分中没有洗下东西?
[align=left]李军芳、谷小会/灰分是指在规定的条件下,试样被灼烧后,所剩残留物经煅烧所得的无机物。煤焦油中灰的主要来源包括:从煤中夹带的灰分,煤焦油在输送和储存过程中进入的灰尘或其他杂质,以及管道或设备等因腐蚀而产生的铁锈等杂质。灰分是煤焦油的质量以及加工利用的重要指标之一,因此煤焦油及馏分油灰分的测定是煤焦油评价必不可少的内容。[/align][align=left] 针对煤焦油及其馏分油的性质特点,参考现行国标“GB/T 508-1985 石油产品灰分测定法”和“GB/T 29748-2013 煤炭直接液化 液化残渣灰分的测定方法”,进行煤焦油及其馏分油灰分的测定。[/align] [b]方法概要[/b]用无灰滤纸作引火芯,点燃放在一个适当容器中的试样,使其燃烧到只剩下灰分和残留的炭,炭质残留物在850 ℃马弗炉中加热转化成灰分,然后冷却并称重。 [b] 具体操作步骤[/b]:1) 用已恒重的坩埚或蒸发皿称取适量具有代表性的样品,准确至0. 0002 g,试样量的多少依试样灰分大小而定。2) 将引火芯安稳的立插在坩埚内的煤焦油中,将大部分试样表面盖住。3) 试样燃烧后,将盛有残渣的坩埚移入加热到(550±10)℃的马弗炉中,在此温度下保持1.0 h~1.5 h,直到残渣灰化完全。然后将炉温升至(850±10)℃,并在此温度下保持1.0 h~1.5 h,直至样品灼烧完全。4) 残渣灼烧完全后,将坩埚放在空气中冷却3 min,然后在干燥器内冷却至室温,称量,准确至0.0002 g。5) 在(850±10)℃条件下进行检查性灼烧,每次20 min,直到连续两次灼烧后的质量之差不超过0.0005 g为止,以最后一次灼烧后的质量为计算依据。注意:含水试样,试验前应进行脱水。对于粘稠的试样,在低温电炉上缓慢加热,使试样不溅出,也不从坩埚边缘溢出。 [b]结果计算:[/b]试样的灰分A按下式计算:A=m1/m ×100%式中: m1一灰分的重量,单位为克(g); m —试样的重量,单位为克(g)。 取重复测定两个结果的算术平均值,作为试样的灰分,结果修约到小数点后三位。
不过分析液相如果没有馏分收集器的话,怎么样手动收集馏分?按出峰时间?有没有实践过的?如果本身样品几十毫升,不多,在分析液相10ul可以基线分离下
在别的网站上看到了这样的求助帖“今天刚刚又碰到一个问题,序列和方法都编写好了,样品和流动相也准备好了,仪器平衡了30多后,开始按Start,可仪器自动进样器不动,在通常显示Last Run的地方换成了nRdy Wait:10,10分钟过去了,还是不进样.重新按Start,还是这样,请问这是怎么回事?怎么解决?在一开始打开工作站时,我曾点击了Reset injector,是不是自动进样器就是这样给不动了,怎么恢复呀?”之后有人回答是“是馏分收集器给去掉了,加上馏分收集器,就好了.因为它是方法中的一部分.”‘我们这的质谱也出现了同样的情况,请问怎么加上馏分收集器?或者说还有别的什么原因吗?
制备色谱收集馏分时,一个色谱峰可以全都收集到一管中嘛,还是要分多个管来收集这个峰。如图[img]https://ng1.17img.cn/bbsfiles/images/2019/10/201910121450214358_4557_3957768_3.png[/img]
各位大侠,请教一个问题。用柱色谱冲洗饱和烃可以得到C13-C30的饱和烃馏分,但我想只要C20之前的烃类,以便做更详细的分析,有没有办法做到只收集前面的组分而不要后面的组分的方法啊?谢谢大家!
各位大侠,请教一个问题。用柱色谱冲洗饱和烃可以得到C13-C30的饱和烃馏分,但我想只要C20之前的烃类,以便做更详细的分析,有没有办法做到只收集前面的组分而不要后面的组分的方法啊?谢谢大家!
现在国VI柴油中增加了总污染物含量的的要求和测定方法按照GB/T 33400-2016 《中间馏分油、柴油及脂肪酸甲酯中总污染物含量测定法 》进行检测。哪位大神有该标准,求传一份,谢谢!
我单位使用的是热电ISQ Ultra trace 气质联用仪用于分析柴油中的多环芳烃的含量,色谱柱为两根DM-5ms 0.25-0.25-30m柱子,从一个进样口分别连在质谱和色谱仪上,柴油样品用固相萃取柱分为饱和烃馏分和柴油馏分,溶剂分别为正戊烷和二氯甲烷。使用一年多都挺正常的,不过从一个月前FID色谱图的芳烃馏分的溶剂峰(二氯甲烷)拖尾严重,影响测定结果。但是芳烃的质谱图和饱和烃馏分的质谱及色谱图都没有溶剂拖尾现象。我尝试过一下办法,均无好转。1.从柱的前端截去一节柱子。2.连接色谱和质谱的柱子对调。3.清洗进样口4.加大分流比5.更换新的柱子发几张图让大家看的更清楚些。芳烃馏分http://ng1.17img.cn/bbsfiles/images/2012/08/201208310727_387546_2412756_3.jpeg饱和烃馏分http://ng1.17img.cn/bbsfiles/images/2012/08/201208310731_387547_2412756_3.jpeg进二氯甲烷http://ng1.17img.cn/bbsfiles/images/2012/08/201208310732_387548_2412756_3.jpeg
作分离纯化的跑层析柱再正常不过了,那么柱子下来的馏分怎么合并呢用什么手段检测
现在使用的是安捷伦1260的馏分收集器,制备的时候不接检测器,直接根据时间段来回收馏分。请问大家,这种情况还需要设置延迟校正的体积吗,我没明白延迟校正是校正的从检测器到馏分收集器的管路体积还是检测上的延迟啊
[align=center][b][font=宋体]【仪器心得】[/font][font=宋体] [font=宋体]馏分收集器[/font] [/font][font=宋体]使用心得[/font][/b][/align][align=center][b][font=宋体] [/font][/b][/align][font=宋体][color=#333333]今天和大家分享一款[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]上面的一部分组件,馏分收集器[/color][/font][font=Arial][color=#333333]1290 Infinity II[/color][/font][font=宋体][color=#333333]。[/color][/font][font=宋体][color=#333333]先和大家简单描述一下什么是馏分收集器,可以收集和定时,在实验室等进行[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]层分析时,仪器能自动计时、计滴分量采样收集层析液,从而代替手工操作实现自动采样收集,提高收集质量和工作效率。我们平时主要用于维生素检验中的样品收集。[/color][/font][font=宋体][color=#333333] [/color][/font][font=宋体][color=#333333]1.关于仪器的使用经验:[/color][/font][font=宋体][color=#333333][font=宋体]每台设备的配制不一样,这台设备是紫外检测器链接的馏分收集装置。在此以维生素[/font][font=宋体]D为例,给大家分享半制备正相高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]及系统适用性的制备,取维生素D标准溶液于试管中,40℃氮吹。残渣用正己烷振荡溶解。取该溶液100μL注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]中测定,确定维生素D保留时间。根据正相系统适用性维生素D的保留时间收集待测液维生素D馏分于试管中。将试管置于40℃水浴氮气吹干,取出准确加入甲醇,残渣振荡溶解,即为维生素D测定液。这就起到了分离的作用。[/font][/color][/font][font=宋体][color=#333333] [/color][/font][font=宋体][color=#333333] [/color][/font][font=宋体][color=#333333]2.仪器的优点和不足[/color][/font][font=宋体][color=#333333]自动收集,能够任意选择,换管定位精确,收集换管不偏离。收集时间,倒、顺定时任意选择,自由互换同规格试管盘,不需要对号就位。能够对参数进行任意设定,对首管、末管进行设定。手工操作被替代,使工作效率得到了提高。但是我们先用的经常会出现一次样品收集到两个试管的问题且一直没有解决。[/color][/font][font=宋体][color=#333333] [/color][/font][font=宋体][color=#333333]3.总结[/color][/font][font=宋体][color=#333333]在工作效率方面代替了之前手动解馏分的装置,大大的节约了时间成本。但是使用前大家也评估一下设备的利用率。如果不是很高,且不长期使用。建议可以之间采用手动的方法在色谱柱后端直接接受待检样品。[/color][/font]
碳硫分析中,钨助剂、纯铁等的添加没有特定的要求,大家有没有研究过助剂对检测结果的影响或是实验中遇到过相关的问题呢?
混酸:硫酸、乙酸、硝酸精馏分离工艺上注意什么?
碳硫分析仪种类繁多,化验分析原理也不尽相同,应用的范围也有区别,价格相差也很大,现将碳硫分析仪常用的几种分析化验方法归纳如下: 1、 红外吸收法(红外碳硫分析仪):试样中的碳、硫经过富氧条件下的高温加热,氧化为二氧化碳、二氧化硫气体。该气体经处理后进入相应的吸收池,对相应的红外辐射进行吸收,由探测器转发为信号,经计算机处理输出结果。此方法具有准确、快速、灵敏度高的特点,高低碳硫含量均使用,采用此方法的红外碳硫分析仪,自动化程度较高,价格也比较高,适用于分析精度要求较高的场合。 2、 电导法(电导碳硫仪):这是根据电导率的变化来测量分析碳硫含量的一种方法,被测样品经高温燃烧后产生的混合气体,经过电导池的吸收后,电阻率(电导的倒数)发生改变,从而测定碳、硫的含量,其特点是准确,快速、灵敏。多用于低碳、低硫的测定。 3、 容量法(气容碳硫仪):常用的有测碳为气体容量法,测硫为碘量法、酸碱滴定法。特别是气体容量法测碳、碘量法定硫,既快速又准确,是我国碳、硫联合测定最常用的方法,采用此方法的碳硫分析仪的精度,碳含量下限为0.050%,硫含量下限为0.005%,可满足大多数场合的需要。 4、 滴定法(滴定仪):非水滴定仪系采用酸碱滴定法测定钢铁碳、硫元素之用。与电弧燃烧炉匹配,适用于一般化验室、炉前化验等使用。 5、 重量法(碳硫联测定仪):常用碱石棉吸收二氧化碳,由“增量”求出碳含量。硫的测定常用湿法,试样用酸分解氧化,转变为硫酸盐,然后在盐酸介质中加入氯化钡,生成硫酸钡,经沉淀、过滤、洗涤、灼烧,称量最后计算得出硫的含量。重量法的缺点是分析速度慢,所以不可能用于企业现场碳硫分析,优点是具有较高的准确度,至今仍被国内外作为标准方法推荐,适用于标准实验室和研究机构。
请问Agilent G1364A/B/C/D 馏分收集器有什么区别现在只知道G1364B(制备级型馏分收集器)和 G1364C(分析级馏分收集器),那么A和 D 呢? 谢谢!
我想把所有样品瓶的同一个组分收集到馏分收集器的同一个试管里,而不是一个样品瓶对应一个试管这样收集,请问应如何设置收集器,希望各位大师指点,多谢啦~
安捷伦馏分收集器重置的时候发生故障,机械臂初始化失败,冒红灯,在故障排除里面说要进行自动化校准操作,实在不知道如何是好?求大佬指路
在所有碳硫分析中,分析结果波动较大我认为都与高频燃烧有关。助熔剂和坩埚的质量是首要关注的内容。特别是助熔剂的燃烧效果直接反应出分析数据的稳定性和碳硫的有效释放。目前我们在功率可调和具备拟合技术的碳硫分析仪器上可以理想地分析铁矿石、硫精矿、焦炭、钛合金、硅铁、地矿粉、白云石、石灰等等样品。因为我们在有意搜集整理和研究各种材料的熔融特性和分析条件。踏踏实实为分析领域做点实事。希望热爱这一事业的朋友能提供一些特殊样品给我。能不能做好我们可以共同努力、共同进步。兴趣就是最好的老师。握手!
[b]适用标准及适用范围SH0175馏分燃料油氧化安定性测定仪是根据中华人民共和国行业标准的SH/T0175《馏分燃料油氧化安定性测定法(加速法)》所规定的要求设计制造的。适用于按SH/T0175标准规定的方法,用加速氧化法测定中间馏分燃料油的固有安定性能。二、主要性能馏分燃料油氧化安定性测定仪,结构上为水浴,,我公司可以根据用户要求按照需求定做。该仪器数显控温,自动计时,报时,并 配有暗箱。三、主要技术指标1、工作电源:AC220V50Hz,功耗:≤2400W。2、控温方式:数显控温表自动控温。3、控温范围:室温~200℃,4、控温精度:设定温度±0.2℃。5、测温元件:热电阻。6、试样数量:4路,同时可以作4个试样。[/b][align=center] [/align]
我们在工厂进行产品监造时在钢中硫含量业主要求为不得高于0.005%。工厂采用的设备是美国LECO公司生产的碳硫分析仪,在见证试验过程中,我们发现工厂在试验时检测数量时通过有效数字规则确认结果为0.006%,超出规范要求。后为使结果达到要求,工厂在设备校验过程中校验仪器时采用一些方法,使数据达到要求。为此双方产生争议,监造方认为,仪器本身的检出量为10万分之一,即使是在校验过程中应以实际结果为准,不能人为改变仪器的设置,使结果合格。但是工厂拿出了国家标准,认为这种校验误差是符合标准要求的,但我们认为,该仪器灵敏度以远远超出国标要求,应以仪器制造厂提供的校验基准为准,即设备校验误差应与设备本身能力相匹配,请问我们的意见是否正确。







 我要推广仪器
我要推广仪器
 下载APP
下载APP