税光厚:为什么要做脂质组学研究?
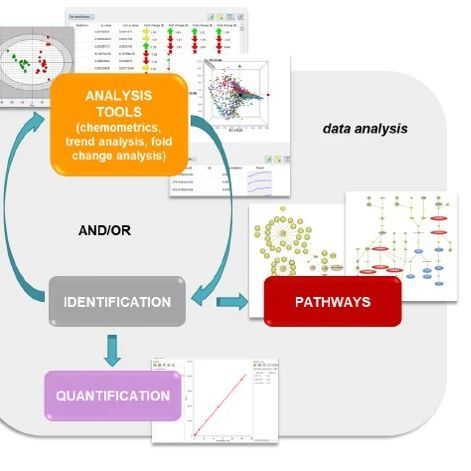
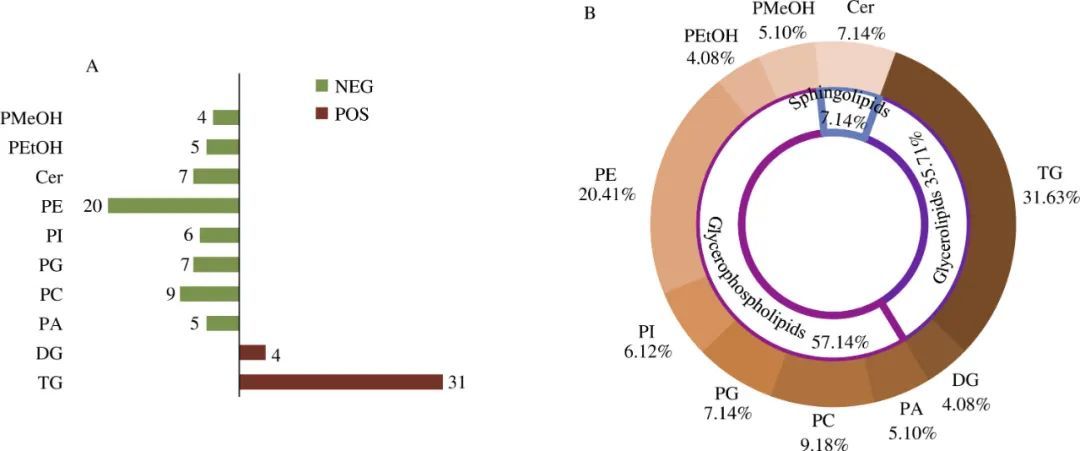
“精准分析”我们的成功故事系列写在前面的话“精准分析,量化释能”——我们的成功故事系列栏目,是SCIEX帮助科学家们分享有意义的科学发现和研究成果的科普平台。我们期待让更多的人感受到科学和“质谱”的力量,“让质谱改变每个人的生活”始终是SCIEX最美好的愿景。SCIEX非常荣幸能够参与到这一系列有趣的科学发现过程中,客户成功就是SCIEX的成功,所以,您读到的是 “我们(SCIEX和客户)的成功故事”。本期科学家税光厚,我们一起谈谈这些年迅猛发展的脂质组学(Lipidomics)。税光厚 研究员个人简介中科院遗传发育所分子发育生物学国家重点实验室研究员,博导,入选中科院"百人计划”;现任中科院遗传发育所所级公共技术中心主任; European J Lipid Sci Technol杂志编委;中国细胞生物学学会细胞代谢分会副会长,中国生物物理协会代谢生物学分会理事,中国病理生理学会内分泌与代谢专业委员会理事,中国抗癌协会肿瘤代谢专业委员会委员 作为大会主席已成功举办四届Lipidall脂质代谢国际会议。长期从事前沿组学技术开发、功能性分子结构鉴定研发、代谢机制相关的研究工作。近年来在Science, Nature, Nature Med., Immunity, EMBO Mol Med, PNAS, Nature Comm,J Clin Invest, J Cell Biol, Redox Biol, Neurobiol Aging, Biochim Biophys Acta, J Biol. Chem, J Lipid Res等期刊发表120余篇关于代谢的论文,发表SCI论文总影响因子约800,被引用6600余次,H-index 45;目前的工作方向包括开发各类生物样品的高通量、超高覆盖率、超高灵敏度脂质组学、前沿代谢组学及代谢流技术,研究代谢调控机制、代谢异常和重大疾病的内在联系等。 什么是脂质组学,脂质组学和代谢组学有什么区别?这个问题的答案来自税光厚老师在科学网blog:gshui的个人博客的置顶博文《脂质组学 vs 代谢组学》。推荐大家关注税老师的blog,通过科学家的视角了解更多关于脂质组学、代谢组学研究的新发现新进展。我们知道,代谢是生物体内各种化学反应的总称,是动植物最为重要的生命活动之一;个体通过各种代谢调节来适应内外环境的变化,是生命活动的基本特征之一。 “代谢组”是生物样本中所有代谢物的集合。代谢物种类繁多(可能超过百万种)、结构多样, 在生物体内不同组织及体液中分布及浓度差异大。主要种类包括:脂类、氨基酸、有机酸、碳水化合物、核酸等。“代谢组学”是系统研究代谢组的一门独立学科,它提供了所有细胞过程的独特生化指纹,可用于鉴定代谢生物标志物,从而阐明潜在的疾病机制,或预测对环境变化或外部干预的反应。“脂质组学” 是系统研究脂质组的一门独立学科, 作为大规模定性和定量研究脂类化合物并了解它们在不同生理、病理条件下的功能和变化的方法学, 能准确全面地提供生物样品在不同生理条件下的全脂信息谱图。从以上定义可以看出,“脂质组学”实际上是“代谢组学”的一个分支。但是,由于脂类代谢(如血浆中约70%的代谢物是脂类) 是动植物的代谢中第一大类,是动植物代谢研究中最为关注的热点,参与能量运输、细胞间的信息通讯与网络调控等生长发育过程中的必需事件。作为细胞膜和脂滴的主要组成成分,各种结构的脂类在广泛的生物学过程,如信号传导、运输作用以及具有不同生化性质的生物大分子分选过程中,扮演着重要角色。 随着近年来脂质组学迅猛发展, 科学家们就逐渐将脂质组的研究即“脂质组学”从“代谢组学”中单独划分出来,现在我们所指的“代谢组学”一般就不再包括系统的脂质组分析了。通过脂质组学研究,阐释脂质代谢相关疾病的发病机理“脂质是细胞膜和脂滴的主要组成成分,在广泛的生物学过程,如信号传导、运输作用以及生物大分子分选过程中扮演着重要角色。对脂质的全面分析, 即脂质组学是了解细胞变化过程中脂质细微的动态变化以及致病机理的先决条件。脂质代谢紊乱和生殖发育缺陷及多种重大疾病如糖尿病、心血管疾病、脂肪肝、肥胖、癌症、老年痴呆等重大疾病密切相关。脂质组成分的变化可直接反映生物体的生理和病理状态,分析组织器官生长发育过程中脂质谱纹的变化及解析相关代谢调控网络机制具有重要的生物学意义和临床价值。税光厚老师领衔首创开发了基于MATLAB脂质组学领域的非靶向数据分析方法与软件(FASEB J, 2006 J Lipid Res, 2007),带动和促进了非靶向脂质组学的发展。 非靶向脂质组学对于挖掘究生物样品的质谱分析中低丰度离子的显著变化、尤其是发现未知代谢物中具有不可或缺的作用,但在定性准确性及效率与定量方面仍面对巨大挑战,并不适用于临床大规模应用或系统生物学大样本分析。税光厚老师团队运用SCIEX QTRAP质谱开发了一系列适用于生物样本的多种物种的高覆盖脂质组的快速靶向定量分析模式(EMBO Mol Med, 2012 PNAS, 2013 J lipid Res, 2014 Neurobiol Aging, 2014 Redox Biol, 2017, Anal Chim Acta,2018),可对全面的脂质库进行准确、可靠的定量分析, 并成功应用于一系列生物医学的基础和临床的重要研究中(PNAS, 2009, 2012, 2018, 2019;JCI, 2011 Nature, 2014 Nature Medicine, 2012 Molecular Cell, 2012 2014 Immunity, 2013 Cell Res, 2016 Development Cell, 2017 Science, 2018 EMBO J, 2018 Nature Comm., 2018);近10年来已经发表100余篇SCI论文,总影响因子超过800。基于LC-MS/MS建立定量脂质组分析方法的一般工作流程(Lam SM, Tian H, Shui G*. BBA-Mol Cell Biol L 2017脂质组学专刊封面文章)掀起脂质的“神秘面纱”近几年,越来越多研究者认识到除了蛋白质,脂质分子在生命体中也发挥重要作用。脂质,是一类自然界存在的疏水或两性的有机物小分子。由“脂质代谢途径研究计划”(Lipid Metabolites and Pathways Strategy,LIPID MAPS)资助的国际脂质分类与命名委员会提出的脂质分类系统,脂质大体分为八大类:常见主要大类脂质的代表结构式(Lam SM, Shui G*. J Genet Genomics 2013. 脂质代谢专刊封面文章)在剔除双键位置异构体,立体结构异构体和sn-构型异构体情况下,根据常见脂质分子组成模块预测真核细胞内部至少有180,000种脂质类化合物。脂质结构的多样性赋予了其多种重要的物理和生物功能:作为细胞膜的重要组成部分,不仅维持细胞膜结构稳定,并且参与调节包括物质运输、能量转换、信息识别与传递、细胞发育和分化以及细胞增殖与凋亡在内的诸多生命活动过程。美国等发达国家都在国家层面部署了规模性项目脂质组学计划,分别已建立联合型的脂质组学研究机构,其中影响力最大的是上文提到的美国的LIPID MAPS项目,获得美国NIH的多期支持的研究项目,取得了非常有影响的成果。我国尚未开始开展类似的注重于联合型的规模性的研究,但近年来众多科研院校都意识到脂质组学的重要性,纷纷组建了各具特色的脂质组学平台,促进了我国脂质组学的发展。脂质组学的一般研究流程脂质组学分析流程主要包括生物样本中脂质的提取、分离分析以及相应的数据分析等。脂质组分析流程(Lam SM, Shui G*. J Genet Genomics 2013. 脂质代谢专刊封面文章) 质谱分析技术助力脂质组学研究应对挑战,奋勇向前探索未知质谱技术的进步极大地推动了脂质组学的快速发展,朝着准确定性、定量的最终目标迈进,这是脂质组学作为一门新兴组学技术分支不断增长和扩展的关键决定因素。脂质组学出现及发展推动了脂质分析平台的研发,特别是在质谱技术进步,已经使众多脂质分子分析成为了可能,但在未来脂质组发展依然有如下挑战需要面对:◆ 如何在缩短分析时间的同时,最大限度地提高脂质覆盖率和定量准确度;◆ 如何实现复杂脂质同分异构体(包括复杂脂质脂肪酸链的双键精确位置)的准确定性与定量; ◆ 如何通过以下途径,从脂质组生成的大数据中获得有生物学意义的模式:以代谢通路为导向的分析方法涵盖连接脂质和非脂质代谢物关键代谢分支定制的生物信息学方法,实现直接和快速代谢通路波动数据可视化等。脂质组定量准确度的转变(Lam SM, Tian H, Shui G*. BBA-Mol Cell Biol L 2017脂质组学专刊封面文章)税光厚课题组火热招聘中——研究人员和博士后如果您对于生命科学有兴趣,热爱科学研究,充满责任感使命感;有代谢组学、脂质组学、液质、气质等方面的研究经验;拥有良好的中英文科学论文写作能力;又刚刚好拥有相关专业博士学位(基本条件)。需要您协助课题组长申请并参与课题研究,并且能够独立申请并开展课题研究任务(基本职责)。
























 我要推广仪器
我要推广仪器
 下载APP
下载APP