求助专业人士,气相色谱在检测有效成分含量很可靠吗?
请大佬教教我 中药中有效成分转移率怎么计算,我只知道是提取液的指标成分含量/药材中指标成分的含量??100%,其中药材中的指标成分含量从哪来的呢?
求HPLC 生姜中有效成分6-姜辣素的含量测量的方法
[table=100%][tr][td]本人做中草药中有效成分含量分析的,用的高效液相,[color=#444444]进行液相分析之前,要对样品溶液进行过滤,周围都用微孔滤头啊, 尼龙的,难道不对?[/color][/td][/tr][/table]
现在在做一批有机磷农药的环境植物样本,因为有效成分含量很低,担心活性炭会吸附有效成分影响检测结果,现在还没有找到合适的脱色方法。除了活性炭以外,还有没有其他好的方法既可以除去色素,又不影响结果
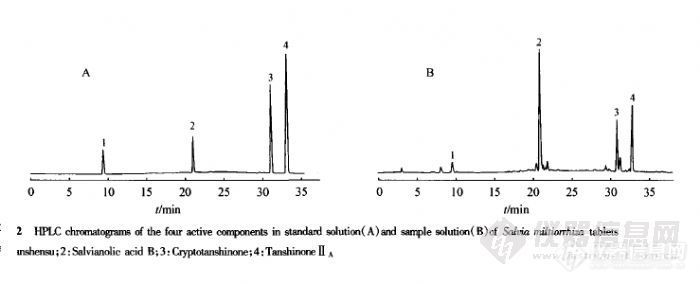
【作者】 孙棣; 刘文英; 梁彩;【Author】 SUN Di,LIU Wen-ying,LIANG Cai Department of Pharmaceutical Analysis,China Pharmaceutical University,Nanjing 210009,China【机构】 中国药科大学药物分析教研室; 中国药科大学药物分析教研室 南京210009; 南京210009;【摘要】 目的:建立一种快速准确同时测定丹参片中脂溶性及水溶性有效成分的方法。方法:建立PR-HPLC法,以甲醇-0.5%甲酸水溶液为流动相进行梯度洗脱,丹参素、丹酚酸B、隐丹参酮、丹参酮ⅡA在Diamonsil C18(250mm×4.6mmID,5μm)柱上得到完全分离。结果:4种物质线性良好,回收率在96.63%~100.3%之间且RSD小于2%。结论:该法能在35min 测定丹参片中脂溶性及水溶性有效成分。http://ng1.17img.cn/bbsfiles/images/2012/07/201207181219_378454_1761902_3.jpg
生药虽来源于植物、动物和矿物,但95%以上来自植物,其所含的化学成分主要是指植物新陈代谢所产生的代谢产物。大多为维持本身生命活动所必需的化合物,这些成分含量较高,而生理活性一般较小,临床应用不多。而植物的次生代谢产物,它们是存在于植物体内的特殊成分,含量较低,但生理活性较强,具有临床应用的价值。通常把生药的化学成分分为三类: 医学教.育网搜集整理 1. 有效成分(active substances) 指具有显著生理活性和药理作用,在临床上有一定应用价值的成分。这类成分仅存在于某些植物中,包括生物碱类、甙类、挥发油类等等,如:利血平(reserpine)是萝芙木降压的有效成分,苦杏仁甙(amygdalin)是苦杏仁止咳平喘的有效成分,薄荷挥发油中的薄荷醇(emnthol)和薄荷酮(menthone)是薄荷辛凉解表的有效成分。 2. 辅成分(adjuvant substances) 指具有次要生理活性和药理作用的成分,有时候,它们在临床上也有一定的应用价值。有些辅成分能促进有效成分的吸收,增强疗效,如:洋地黄皂甙能促进洋地黄强心甙的吸收,从而增强洋地黄的强心作用。有些辅成分能使有效成分更好地发挥作用,如槟榔中的鞣质,可保护槟榔碱(arecoline)在胃液中不溶解,而到肠中才被游离出来,木栓、角质、粘液、色素、树脂等。在生药鉴定、有效成分测定或在制备药剂时必须考虑它们的存在与性质。 3. 无效成分(inactive substances) 指无生理活性,在临床上没有医疗作用的成分。它们包括纤维素、木栓、角质、粘液、色素、树脂等。在生药鉴定、有效成分测定或在制备药剂时必须考虑它们的存在与性质。 上述分类并不是绝对的和固定不变的,应根据具体的生药进行具体分析,才能确定某成分是否是有效成分、辅成分或无效成分。如:鞣质在地榆与五倍子中为有效成分,在大黄中为辅成分,而在肉桂中为无效成分。同时应从发展的观点来分析,随着人们的不断实践,特别是现代科学技术的发展,生药中越来越多的化学成分被认识,用于药理研究,进而被开发用于临床。原来认为是"无效"成分,现在不少已发现了它们的医疗价值,而成为有效成分了。如:天花粉蛋白质有引产、抗癌作用,蘑菇多糖(lentian)对实验动物的肿瘤有明显抑制作用,叶绿素能促使肉芽生长,菠萝蛋白酶有驱虫、抗炎、抗水肿的作用。 医学教.育网搜集整理 生药的化学成分不仅与药理作用、临床应用有密切的联系,而且对于生药的鉴定、质量评价、新制剂的开发研究、新资源的发掘利用均有密切联系。随着化学成分的生源(biogenesis)和生物合成(biosynthesis)研究的深入,对植物新陈代谢及其代谢产物的内涵也将不断充实和发展。
【摘要】 目的考察硫熏干燥对菊花中有效成分的含量影响。方法采用HPLC法测定自然干燥和硫磺熏蒸干燥菊花中绿原酸、槲皮素、木犀草素的含量;采用分光光度法测定菊花中总黄酮的含量;采用盐酸-副玫瑰苯胺法测定各菊花中SO2的残留量。结果与自然干燥的菊花相比,硫熏菊花中的槲皮素、木犀草素和总黄酮含量低于前者,而绿原酸含量高于前者,SO2的残留量远远高于前者。结论硫磺熏蒸对菊花中有效成分的含量变化有一定的影响,建议产地加工时应限制使用硫熏干燥法。 【关键词】 菊花; 硫熏干燥; 绿原酸; 槲皮素; 木犀草素; 总黄酮 菊花是菊科植物菊花Chrysanthemum morifoliumRamat的干燥头状花序,为我国常用中药,具有疏风、清热、明目、解毒之功效。临床上主要用于治疗头痛、眩晕、目赤、心胸烦热、疔疮肿毒等症。目前市售的菊花多采用硫磺熏蒸的方法加以干燥,以达到漂泊、美化外观和防霉生虫等目的。但硫磺熏蒸后的药材是不利于入药的,因为其中残留的亚硫酸盐会对人体造成咽喉疼痛、胃部不适等伤害。此外,熏蒸后产生过多的SO2也会对大气造成严重污染。为考察硫磺熏蒸对菊花中绿原酸和黄酮类化合物含量的影响,今对自然干燥菊花与硫熏菊花中绿原酸、槲皮素、木犀草素及SO2的残留量进行了含量测定研究,为菊花资源的科学应用提供参考。 1 仪器与材料 1.1 仪器戴安UltiMate 3000高效液相色谱仪, UltiMate 3000四元泵,UltiMate 3000variable wavelength检测器,Chameleon工作站,KQ-250B超声波清洗器(昆山市超声仪器有限公司),Spectrum922E型分光光度计。 1.2 材料白菊花采自河北安国祁药标准化种植基地。芦丁对照品(100080-200707)、绿原酸对照品(110753-200403)、木犀草素对照品(111720-200603)、槲皮素对照品(100081-200406)均购自中国生物制品检定所。甲醇采用色谱醇,水为重蒸馏水,其余均为分析醇。 2 方法与结果 2.1 硫磺熏蒸菊花的制备将新采摘的菊花头状花序摊放于直径为90cm的带有支架的尼龙丝网圆筛中,把自制的体积约1.26m3聚氯乙稀罩自上而下套在外面,距地面约为10cm。将适量的硫磺置于陶瓷器皿中,点燃后迅速推入到垂直于圆筛的下方地面上,待其燃烧完全后,将聚氯乙稀罩下调至和地面充分接触,形成密封环境,保存12 h后取出,通风至干,备用。
舒肝丸为医院制剂。主要由龙胆、柴胡、黄芩、栀子(炒)、泽泻、木通、车前子(盐炒)、当归(酒炒)、地黄、炙甘草等中药组成。具有清肝胆,利湿热之作用。用于肝胆湿热,头晕目赤,耳鸣耳聋,胁痛口苦,尿赤,湿热带下。为了控制检测指标,我们建立了舒肝丸三种有效成分龙胆苦苷、栀子苷、黄芩苷的含量测定方法,以期为完善制剂质量控制提供参考依据。摘要: 目的 建立疏肝丸中龙胆苦苷和栀子苷、黄芩苷的含量测定方法。方法 液相色谱法。色谱柱:Kromasil C18柱(4.6mm×250mm,5µm);流动相:甲醇-0.5%冰醋酸溶液(25:75);流速1.0ml/min;柱温:35℃;检测波长:283nm。结果: 龙胆苦苷在0.525~5.25μg范围内呈线性关系(r=0.9998 ,n=6),平均加样回收率为95.6% RSD为1.08% (n=6)。栀子苷在0.635~6.35μg范围内呈线性关系(r=0.9996 ,n=6),平均加样回收率为97.04% RSD为0.58 % (n=6)。黄芩苷在10.658~106.58μg范围内呈线性关系(r=0.9996 ,n=6),平均加样回收率为99.24 % RSD为0.37 % (n=6)结论 该方法操作简便,结果可靠,可用于疏肝丸中龙胆苦苷、栀子苷和黄芩苷的含量测定。关键词:高效液相色谱法;舒肝丸;龙胆苦苷 栀子苷 黄芩苷 1 仪器与试药1.1 仪器岛津LC-20AT高效液相色谱仪,SPD-M20A检测器, LCsolution色谱工作站, Metteler AB265S(0.01mg);Sartorius BS124S(0.1mg)1.2 试药 龙胆苦苷、栀子苷、黄芩苷对照品(中国药品生物制品检定所,批号110770-201013;0749-200007;110715-201016);甲醇为色谱纯,冰醋酸为分析纯,水为纯化水。舒肝丸为医院制剂,批号:20130629 20131102 201402162 方法与结果2.1 色谱条件Kromasil C18柱(4.6mm×250mm,5µm)流动相甲醇和0.2%磷酸(按以下梯度进行洗脱),柱温35℃,流速1.0 mL·min - 1 ,进样量10μL;波长254nm。时间(分钟)流动相(甲醇)流动相(0.2%磷酸)0-252080 25-30 20-4380-5730-50435750-6043 572.2 对照品储备液 精密称取龙胆苦苷、栀子苷、黄芩苷对照品约10mg, 置100ml量瓶中,精密称定,加甲醇制成每1ml含0.1024、1034、1058mg的对照品溶液。2.3 供试品溶液的制备 取本
根据GB2023-80来检验乳酸的有效成分,在标准中,使用过量的氢氧化钠来中和,再用强酸硫酸来反相滴定的。如果用1当量浓度的浓硫酸来滴定,计算公式为乳酸的含量是(V2-V1)×N×0.09008/g×100。如果是用0.6028摩尔浓度的浓硫酸来滴定,这条公式怎么变换呢?如果是同样摩尔浓度的盐酸来代替硫酸可以吗?各位大侠,化学学得不精,就麻烦各位帮忙想想了。先叩谢各位了。
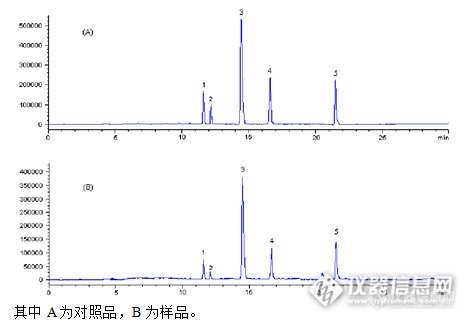
中药有效部位新药5个主要成分的含量测定方法 有效部位指从植物、动物、矿物等物质中提取的一类或数类成分和一种或数种有效成分组成的或仅由一类或数类成分组成的有效部位及其制剂。其有效部位含量应占提取物的50%以上,并对每类成分中的代表成分和组成的有效成分进行含量测定且规定下限,条件许可的也规定上限,有毒性的成分必须增加上限控制。有效部位中药之所以应该得到发展和重视 ,这和它的优势是分不开的:有效部位中药坚持中医药理论的指导,做到中药的基础研究和给药途径、剂型相结合;有效部位中药的药效比传统中药提高,改革后的新剂型,比原有剂型一般在药效上有所提高;疗效确切,服用安全可靠 ,毒副作用低;组成有效部位中药的药物物质基础或药效基础应基本得到明确;具备完善可控的质量标准。 仪器与试药: 对照品1、对照品2、对照品3、对照品4、对照品5均购自中国药品生物制品检定所,中药有效部位片(自制)、乙腈和甲醇均为色谱纯,水为娃哈哈纯净水,其余试剂均为分析纯。 Agilent1200系列快速分离液相色谱仪,包括在线脱气机,四元泵,高性能自动进样器,柱温箱,G1969A 单重四极杆质谱仪,配有 ChemStation色谱工作站;安捷伦6210高分辨飞行时间质谱仪,配有标准电喷雾离子源(ESI),及Masshunter 工作站和AnalystQS 质谱分析软件;SB3200-T 超声发生器;METYLER AE240型十万分之一电子天平。 TOF/MS质谱条件 采用ESI离子源,考察了质谱条件毛细管电压,雾化气压力,干燥气流速及干燥气温度等对质谱响应的影响,正离子模式下选择性、灵敏度及检测信号等优于负离子模式,最终确定质谱分析参数如下:正离子模式下(ESI+):毛细管电压4000V,雾化气压力40psi,干燥气流速10L/min。,干燥气温度 350℃,碎片碰撞电压范围 100~425 V;;质量数扫描范围 m/z 100~1500。色谱柱:Agilent Zorbax XDB-C18(2.1×50mm,1.8μm);流动相:A相为水(0.1 %甲酸),B相为乙腈,梯度洗脱;梯度洗脱程序如下:0~10 min, 5~23% B; 10~20 min, 23~23% B,20~25 min, 23~45% B; 25~30 min, 45~95% B;进样量:1μl;进样前以流动相梯度初始条件平衡10 min;流速:0.2 mL·min[f
今天去抽检,厂家说益母草颗粒剂的有效成分随着时间变长,含量也降低。是什么原因呢,难道没有办法解决这个问题吗?
在研究中药时,往往会提到有效成分和活性成分,那么这两个是不是一个概念呢?
主要研究内容 (1)化学研究:从平喘作用较强的艾叶油中沸点部位,经真空精馏,柱层析,TLC制备,GC~MS分离和鉴定了18个成分,其中有4个具有较强的平喘作用,即3个含氧单萜(萜品烯醇~萜4,反式~葛缕醇,α~萜品烯醇),1个倍半萜(β~石竹烯)。 (2)药理研究:发现艾叶油及其有效成分对豚鼠气管平滑肌均有较强的直接松弛作用。各有效成分的作用均较艾叶油为强,以ED50与EC50进行比较,分别强4~30倍。艾叶油及其有效成分的小白鼠LD50均在1000~2000mg/kg。艾叶油及α~萜品烯醇家兔每日以相当于成人剂量25倍灌胃,连续20~30天,主要脏器的功能和结构无明显毒性。 (3)临床部分:艾叶油及其有效成分治疗慢性支气管炎和支气管哮喘共2000余例患者,均有较强的临床治疗作用。艾叶油胶丸、气雾剂、β~石竹烯胶丸、萜品烯醇~4胶丸口服或气雾吸入治疗慢性支气管炎,疗程20天,总有效率78.53~100%,控显率33.51~56.37%,对咳、痰、喘、哮四症均有不同程度疗效,尤以喘、哮为佳。在治疗过程中无明显毒副反应。 主要完成单位 浙江省中医研究所 浙江医科大学 上海医药工业研究所
农药有效成分的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]快速分析方法 -------------------------------------------------------------------------------- 上传时间:2005年6月18日 点击:430 农药有效成分的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]快速分析方法 第1部分 十二种农药Fast gas chromatographic method for analyzing active ingredients of pesticides-Part 1:12 kinds of pesticides1 范围本标准规定了十二种农药(见附录A)有效成分的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]快速分析方法。本标准适用于具有高灵敏度热导池检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],对适合于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的农药进行快速定性及定量分析。2 引用标准下列标准所包含的条文,通过的本标准中引用而构成为本标准的条文。本标准出版时,所示版本均为有效。所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。GB/T 1605-79(88) 商品农药采样方法GB/T 4946-85 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法术语JB 5225-91 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测试用标准色谱柱3 试验方法3.1 方法提要试样用丙酮溶解,根据不同有效成分,选择联苯或邻苯二甲酸二甲酯、邻苯二甲酸二丁酯、邻苯二甲酸二正戊酯、邻苯二甲酸二正辛酯等为内标物,在2%OV101、10%OV17色谱柱上进行色谱分离;相对保留值定性,相对质量响应值Sm用于定量测定。3.2 抽样按GB/T 1605执行。3.3 试剂和溶液。3.3.1 丙酮:分析纯。3.3.2 三氯甲烷:分析纯。3.3.3 农药标样或定性工作标样(即已知定性农药样品)。3.3.4 内标物:已知准确含量,无干扰分析的杂质。3.3.5 固定液:甲基硅油OV101和苯基(50%)甲基聚硅氧烷OV17。3.3.6 载体Chromosorb W AW DMCS 150~170 μm。3.4 仪器GB/T 165787-1996[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:具有高灵敏度热导池检测器,灵敏度S≥2000mVmL/mg(苯),噪声≤30μV,飘移≤0.1V/0.5h。色谱数据处理机或满量程2mV的记录仪。色谱仪:a)1m×2mm(id)不锈钢柱或玻璃柱。柱填充物为OV101固定液涂在Chromosorb W AW DMCS载体(150~170μm)上,固定液:载体=2:100(m/m)。b)1m×2mm(id)不锈钢柱或玻璃柱。柱填充物为OV17固定液涂在Chromosorb W AW DMCS载体(150~170μm)上,固定液:载体=10:100(m/m)。 3.5 色谱柱的制备按JB 5225标准制备。3.6 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]操作条件。见附录B3.7 测定步骤3.7.1 农药标样溶液的配制根据附录D计算农药标准物质的称样量m(g)=0.05/Pi,称样量应约等于计算值,称准至0.2mg,置于10mL容量瓶中,式中Pi为农药标准品质量百分含量。 miPi计算内标物的称样量wa(g)= ————— ,称样量应约等于计算值,称准至0.2mg,置于上 KPa 述同一容量瓶中。Pa为内标物的质量百分含量。农药峰面积与内标物峰面积相等时K=纯农药质量/纯内标物质量。用丙酮溶解并稀释至刻度,摇匀。3.7.2 农药定性工作标样溶液的配制在没有农药标样的情况下用已知农药定性工作标样三滴加内标物一滴(固体加少许)于同一称量瓶中用两酮溶解并稀释,摇匀,做定性测定使用。3.7.3 农药试样溶液的配制计算农药样品的称样量为mi=0.05/X(g)(X为农药样品的标示含量),称样量应约等于计算值。标准至0.2mg置于另一个10mL容量瓶中。 miX计算内标物的称样量应为mi(g)=————— ,称样量应约等于计算值,称准至0.2mg,置于 KPa上述同一个10mL容量瓶中,用两酮溶解并稀释至刻度,摇匀。 3.7.4 相对质量响应值Sm的测定在附录B规定操作条件下,待仪器稳定后注入数针配制好的农药标样溶液。待相邻两针的峰面积比值基本稳定后,分别按1μL,2μL,3μL,4μL,5μL五种不同的进样量进样,每种平行5次。3.7.5 农药试样定量测定按附录B规定的操作条件,待仪器稳定后注入数针配制好的农药试样溶液,待相邻两针的峰面积比值基本稳定后,重复进样3~5次,进样量应与所测值SM的进样量一致。 3.7.6 色谱柱死时间tM的测定按附录B规定的操作条年,待仪器稳定后分别向两根色谱柱中注入1μL空气,记录氮气出峰时间tM。3.7.7 没有农药标准品时的定性测定按附录B规定的操作条件,待仪器稳定后分别向两根色谱柱中注入农药定性工作标样溶液,并记录农药i和内标物s的保留时间。注意农药定性工作标样峰面积要与被测农药试样峰面积相接近。3.8 计算3.8.1 定性计算按式(1)分别计算农药标样(或农药定性工作标样)和农药试样i对内标物s的相对保留值ri,s tR(i)-tMri,s= ————— tR(m)-tM 式中:tM——色谱柱的死时间,min;tR(i)——农药i的保留时间,min;tR(m)——内标物的保留时间,min。3.8.2 定量计算3.8.2.1 相对质量响应值Sm的计算。按式(2)计算农药标样i对内标物s的相对质量响应值Sm。按附录E荻克逊准则进行统计检验剔除可疑值后分别求出进样量1μL,2μL,3μL,4μL,5μL的S=平均值。注:热导池检测器的相对质量响应值Sm每年需用农药标准品检定一次,仪器检修或更换部件后应重新用农药标准品测定Sm值,以保证定量数据的准确。 AimsPaSm=—————— …………………………(2) AsmiPi式中:Ai——农药标样i的峰面积值,mm2或μVs;As——内标物s的峰面积值,mm2或μVs;mi——农药标准品i的质量,g;ma——内标物s的质量,g;Pi——农药标准品i的质量百分含量,%;Pa——内标物s的质量百分含量,%。3.8.2.2 农药样品含量的计算按式(3)计算农药样品百分含量X,按附录E荻克逊准则进行统计检验剔除可疑值后,求出平均X值。 Aims'Pa'X =—————— …………………………(3) As'mi'Sm 式中:Ai'——农药标样i的峰面积值,mm2或μVs;As'——内标物s'的峰面积值,mm2或μVs;mi'——农药标准品i'的质量,g;ms'——内标物s'的质量,g;Pa'——内标物s'的质量百分含量,%;Sm——农药有效成分对内标物的相对质量响应值。4 判断原则4.1 定性判断比较农药样品与农药标准品(或农药定性工作标准样品)i对内标物s的相对保留值ri,s在两根柱中是否都相同,误差不超过附录D相对保留值平行偏差,则判断农药样品为i农药,否则为假农药。4.2 定量判断将定量计算结果与农药i的产品标准规定的含量或农药标签标明的含量相比较,不超过附录D中定量平行偏差的为合格农药。4.3 仲裁判断按农药产品标准规定的分析方法进行。
5月30日,从国家知识产权局获悉,由中国科学院武汉植物园科研人员袁晓、袁萍共同发明的“一种从紫草中分离出萘醌类有效成分的方法”获国家发明专利授权。(专利号:ZL 201110164992.8) 紫草为紫草科植物新疆紫草Arnebaieuchroma (Royle) Johnst的干燥根,紫草中主要药效成分为萘醌类色素,包括紫草素、去氧紫草素、β,β二甲基丙烯酰紫草素、乙酰紫草素等。从紫草中分离出萘醌类有效成分,其药理作用有抗病原微生物、抗病毒、抗炎和消化道平滑肌等作用。主要具有抗癌作用,由新疆紫草中提取的紫草素(Shikonin),510mg/kg可完全抑制腹水型肉瘤180细胞的生长。10mg/kg可延长带瘤小鼠生命92.5%。紫草素(Shikonin)对小鼠肝癌H22和Lewis肺癌的放射增敏作用。 目前,对紫草萘醌成分的提取纯化方法有较多的研究报道,但大多是传统的提取分离工艺,分离时间长,提取杂质多,溶剂耗损大,不易分离得到高纯度的药用成分。 本专利提供的提取分离方法易行,操作简便,其方法是利用Dr FlashⅡ型中低压制备色谱,高效分离抗癌活性成分的优化工艺, 能获得纯度较高的有效成分,且含量在95%以上。应用该专利技术提取的活性成分不仅有抗肿瘤的作用,还有抗炎、解热、镇痛、镇静、抗体病原微生物、抗生育及保肝作用,能广泛用于医药卫生行业。
超滤法。刘荣华等对大孔吸附树脂提取胆红素的工艺进行了考察,在应用CDA-40型大孔吸附树脂、 pH值为5~6、吸附剂用量为4g/10mL胆汁、硫酸铵盐浓度70%、搅拌吸附时间为4h的条件下,胆红素的提取率达85%以上,纯度达93%,且工艺简便、大孔吸附树脂再生容易。陈延清用7种不同种类的大孔吸附树脂来精制乐脉胶囊,用HPLC法测定丹参素、芍药苷的含量,结果显示,用树脂精制后提取物的含固率显著降低,丹参素的损失很大,芍药苷在部分树脂的保留率低于80%。 5 结语 大孔吸附树脂在天然药物的分离、富集方面有着广泛的应用前景,并日益显示出其独特的作用。目前在天然药物化学成分分离方面最常用的树脂有D-101,DA-201,AB-8,H103,LD605, CDA-40, D1300型等,还有NKA和SIP系列。目前大孔吸附树脂在苷类成分分离方面应用较广,在其他类化学成分的分离方面应用研究有待深入。应用大孔吸附树脂可将天然药物的有效成分分离出来,特别有利于解决天然药物大、黑、粗的问题。随着在天然药物化学成分提取、分离、富集中的进一步应用,大孔吸附树脂必将有利于天然药物制剂工艺的改进,有利于促进天然药物现代化研究的进程。
农药登记上经常要用到,有效成分的波谱特征,ultraviolet, visible, infrared etc., 请问有没有人做过这种总结呢,把农药有效成分的波谱特征都总结出来。或者网上那里可以查询呢?或者什么化工出版社有没有出过类似的书呢,能否推荐推荐
活性成分和有效成分的定义以及它们的区别
大家好!请问有谁知道苦瓜提取物中有哪些有效成分
各位大侠: 请帮忙找一下这些农药的有效成分是什么:高保七品红金宁大拇指杀菌优大康
我这里有一蒙药方,属碳化灰。有效成分可溶入水中,疑似无机盐,如何提取其有效成分?谢谢!我的邮箱bjh_782163.com 多谢指教! [em0808]
谁能提供一些防风中有效成分[b][color=#f10b00]升麻素苷[/color][/b]和[b][color=#f10b00]5-O-甲基维斯阿米醇苷[/color][/b]的具体资料?
导语:近日,中美研究人员在中药领域又有新的研究发现,他们从传统中药材延胡索(又名元胡)中找到并确认了一个新的镇痛活性成分去氢紫堇球碱(DHCB),以此为基础或许可研制出新型止痛药,研究的发现每次都很鼓舞人心,这不仅是医药科技的发展,同时也验证了我国传统中医药的有效性。但是这些研究发现也恰恰正揭露了我国大多数中药面临的最严峻的问题--中药标准化。毕竟,还有许多的中药药效成分不明确,成分含量、质量标准不健全。我们在为中药材领域的新研究发现兴奋激动的同时,我们也应该意识到放在我们面前的任务有多么艰巨,我们需要更加努力,明确更多中药的有效成分,制定合理的中药标准,才能把我国中医药发扬光大。 其实除了本次在元胡中发现的镇痛成分之外,以往类似的发现还有很多,如蜈蚣的中药成分可做镇痛剂,中药山茱萸活性成分能够保护海马神经元,中药迷迭香成分对视网膜具有保护作用等等,下面我们就来详细了解一下研究人员对中药活性成分研究的一些情况。 中美研究新发现一传统中药的镇痛成分 中国科学院大连化学物理研究所与美国加州大学欧文分校研究人员合作,新发现了延胡索(又名元胡)中的镇痛活性成分去氢紫堇球碱(DHCB)。动物实验显示,它对慢性疼痛可能有很好疗效,并且没有耐药性。而吗啡等阿片类镇痛药,虽然开始药效很强但很快就会产生耐受,需要不停加大剂量才能达到相同治疗效果,耐药性与作用持续时间不及DHCB。以此项研究为基础或许可研制出副作用小、无成瘾性的止痛药。 NRR:中药山茱萸活性成分能够保护海马神经元 研究发现炮制过的山茱萸抗衰老作用明显,尤其是抗脑部衰老,其活性成分5-羟甲基糠醛对H2O2损伤的大鼠海马神经元具有一定保护作用,可以提高损伤细胞中超氧化物歧化酶的活力,减轻H2O2引起的神经细胞的损伤。 南京中医药大学Mingyan Wang教授带领的团队首次通过细胞生物学和分子生物学手段,探讨山茱萸活性成分5-羟甲基糠醛对氧化损伤的大鼠海马神经元保护作用的分子机制。 PNAS:蜈蚣的中药成分可做镇痛剂 蜈蚣是常用的动物中药药材,用于治疗中风和疼痛等疾病。中国科学院昆明动物研究所赖仞研究员领导的研究团队近期对蜈蚣来源的活性多肽及其发挥药效的作用机理进行了较为系统的研究,发现了一系列作用于离子通道的蜈蚣神经毒 IOVS:中药迷迭香成分对视网膜具有保护作用 草药在亚洲整个历史及早期欧洲文化中被广泛使用。最近几年,草药受到了西方医学的重新关注。目前,科学家们正从许多药材中分离提取活性化合物,并记录它们的抗氧化及抗炎症作用。在最近发表于IOVS杂志上的一项研究中,美国桑福德-伯纳姆医学研究所的Stuart A.Lipton博士及其同事们报告称,草药迷迭香中的一种组分――鼠尾草酸(carnosic acid)能够促进眼睛的健康。 我国研发投入严重不足 中药国际化困难重重 通过上述例子的介绍,大家可以发现,虽然对中药成分的研究不断有新的成果出现,但是相对于我国博大精深的传统中医药来说还是九牛一毛。同时我们也可以发现,许多研究都是我国与国外专家合作研究,甚至有许多是国外科学家进行的研究。这种情况应该引起我们的重视,中医药是古人流传下来瑰丽的知识遗产,在实现中医药国际化,标准化的道路上我们作为炎黄子孙有责任也有义务去做好相关的研究工作。但现实反映出我国的研发投入严重不足,这使得我国中药国际化的路程漫长而艰辛。 我国中医药产业发展水平仍然偏低,研发投入严重不足,仿、改制品种泛滥以及缺乏标准和规范等已成为主要制约瓶颈。由于缺少受市场认可的中药标准体系,中医药在国际市场上处于尴尬地位,身份难以合法化,难以在国际市场上成为主流。面对这样的现状,我国更应该加大中药研发方面的投入,使更多的中药成分及作用机制得以被发现。 中药标准体系的缺失 有效成分确定困难 就目前而言,中药标准体系的缺失一直是制约中药行业发展的重要因素。尤其在半成品加工环节,无法通过准确的检验指标来确定有效成分的含量。目前,中药的主要检测方法还是依赖人工检测,就是有经验的专家通过观察、品尝等办法判断原材料的好坏,至于有毒添加物,更是没有相关标准和成熟的检测办法。此外,中药行业要开拓国际市场,中药就要融入西方的医药体系,尤其要适应其在从实验论证到具体应用都充满量化、具体化标准的习惯时,便会遭遇文化差异。 不过,中药标准体系的建立,也不是一件容易的事情。传统中药复方成分不明,因此给中药复方的质量控制、作用机制研究带来极大的困难;中药的药效、毒理、制剂等也无法向现代化学药看齐。简单来说,原先是把一些药材按照比例配在一起,但要按照中药行业标准执行,就需要先提取这些药材中产生作用的物质,再把这些物质按照规定的比例融合在一起。制定中药行业标准先决条件是要分析每种药材提取的各种物质并进行分门别类。 结束语:我国中药标准的缺失大大制约了我国中医药的国际化发展,面对种种困难,我国应该加大中药研发投入,加强专项研究,尽早尽快的明确中药的有效成分、作用机制,这将是我国中药产业未来应该努力的方向,同时也希望中药企业加强自律,此前,出现很多关于我国中药原材料掺假的报道,给我国中药企业敲响了警钟。希望通过各方共同努力,能使我国中医药在国际上绽放光彩。
不知道提取物中有效成分含量,想测定其含量,我这个标准品配置多大浓度,我怎么确定呢?这个是不是得试验呢?
请问减肥香皂的有效成分是什?
[color=#444444]混合物中含有大概4~5种成份,这几种成份基本上都知道是什么,而其中的有效成分含有的基团其他成份中均不含,一般红外都是用来做定性测量,所以想问下能否通过这种方法来进行定量测量,精度上要求不是很高,如果能的话,测试有什么具体的条件么[/color]
中药有效成分提取分离技术研究进展
有效成分是什么?有没有活性碳?
中药有效成分常见色谱前处理方法第一步:提取法: 用相应的溶剂使有效成分溶解出来的,溶剂根据有机物的性质来选择。可以冷浸,如果稳定性允许,为加速溶解的过程,可采用超声处理,加热回流(包括索氏提取)。选择适当的溶剂可以减少杂质。基本原则是能用纯的甲、乙醇不用带水的,能用非极性的不用极性的,因为消除极性杂质一般比非极性的麻烦。根据相似相溶的原则,非极性溶剂溶解的极性物质较少。 蒸馏(多为水蒸气蒸馏)也较多使用,同时具有提取和一定的分离作用。 某些物质还可以用升华法:樟脑,咖啡碱(178℃以上就能升华而不被分解)等。升华法易产生挥发性的焦油状物,粘附在升华物上,不易除去,另外,升华不完全,甚至会分解,不能用于含量测定。 第二步:分离和纯化: 1、液液萃取 基本上都是水溶后,用乙酸乙酯、乙醚、氯仿、石油醚等与水不相溶的有机溶剂。一般分析的量较少,微溶于水的物质都可以用液液萃取的方法。通过合适的酸化/碱化步骤,可有效的去除杂质。 2、沉淀 利用重金属等与待分析物形成络和物或难溶盐沉淀而分离,然后再用酸、碱等把它还原出来。铅盐常用以沉淀有机酸、氨基酸、蛋白质、粘[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]、鞣质、树脂、酸性皂甙、部分黄酮等。例如含绿原酸的溶液加醋酸铅溶液,搅拌使沉淀,离心,弃去上清液,沉淀物加5%硫酸溶液,用醋酸乙酯萃取。常用雷氏铵盐使生成生物碱沉淀析出。橙皮甙、芦丁、黄芩甙均易溶于碱性溶液,加入酸后可使之沉淀析出。某些蛋白质,可以变溶液的pH值利用其在等电点时溶解度最小的性质而使之沉淀析出。此外,还可以用明胶、蛋白溶液沉淀鞣质;胆甾醇也常用以沉淀洋地黄皂甙等。 3、析出法:包括盐析和改变溶剂极性析出。 盐析法是在水溶液中、加入无机盐,可使某些成分在水中的溶解度降低沉淀析出,而与水溶性大的杂质分离。常用作盐析的无机盐有氯化钠、硫酸钠、硫酸镁、硫酸铵等。三七皂甙乙、掌叶防己碱,小檗碱等都可以用种方法。不过,一般分析上是在水溶液中加入一定量的食盐,再用有机溶剂萃取,例如麻黄碱、苦参碱等水溶性较大的物质。 改变溶剂极性析出是在溶液中加入不同极性的溶剂是物质析出。就像生产中水提醇沉、醇提水沉。 4、柱色谱法: 常用的固定相有硅胶、氧化铝、聚酰胺、大孔树脂、活性炭、硅藻土等。柱色谱法有两种,是简单的吸附解吸,理论塔板数相当于一,跟萃取差不多,也可称富集,例如像现在用得很广的SPE(solid phase extract)。另外一种则是像色谱先驱Tswett那样,样品加在柱上形成一窄带,然后再分步或连续洗脱。 第一种较简单,活化后,按要求预处理,然后让样品溶液通过吸附剂,吸附饱和后,根据不同物质的解吸条件,选择不同吸脱溶剂吸脱下来。关于SPE小柱的使用这里不介绍了,仅举一更实际的例子:取天麻细粉5g,加甲醇50ml,水浴加热回流1小时,过滤,滤液蒸干,残渣加水30ml溶解,用氯仿萃取2次,每次10ml,弃去氯仿液。水液加热挥去残余氯仿,加活性炭(不活化)2g,微沸20分钟,放冷,加硅藻土1g,搅拌均匀,用滤纸过滤,再以40ml水洗涤残渣,滤液弃去;残渣于水浴上干燥半小时,然后置锥形瓶中,加无水乙醇50ml,回流半小时,过滤,滤液浓缩至干,加甲醇1ml使溶解,作为供试品溶液。其实,如果不是含量测定的话,上面的做法可以达到目的。如果是含量测定,那就要用柱子,并确定一下吸附剂的使用量,防止待测物损失。 第二种方法,最重要的是让样品在柱上形成窄带。我们可以近似的认为,窄带的高度相当于理论塔板高度,柱长除以理论塔板高度就是理论塔板数了。实际上洗脱时,待测成分在柱上的实际高度不一定等于窄带的高度。如果用洗脱剂直接溶解样品,就可以直接上柱了。如果不是的话,就要用一定的吸附剂吸附样品,在把样品的溶剂挥干后,在加到柱子。洗脱条件应通过实验确定,包括洗脱溶剂、洗脱体积、洗脱速度。 根据不同的吸附剂,具体的应用不同: (1)硅胶:硅胶颗粒表面有很多硅醇基。硅胶吸附作用的强弱与硅醇基的含量多少有关。硅醇基能够通过氢键作用吸附水分而降低吸附能力,硅胶的吸附性根据其含水量不同分成多个级别。硅胶的活化:100~110℃时,硅胶表面水分即能被除去;500℃时,硅胶表面的硅醇基脱水缩合转变为硅氧烷键,失去吸附性。硅胶是酸性吸附剂,用于中性或酸性成分分离。不活化的硅胶,可利用分配原理分离色素。硅胶又是一种弱酸性阳离子交换剂,其表面上的硅醇基能释放弱酸性的氢离子,所以可作为离子交换剂,分离碱性化合物。吸附硅胶柱的洗脱溶剂成很多,从非极性到极性的顺序洗脱。 (2)氧化铝:氧化铝偏碱碱性,最适宜用于生物碱类的分离,不宜用于醛、酮、醋、内酯等类型的化合物分离。氧化铝用水洗至中性,称为中性氧化铝,中性氧化铝适用于酸性成分的分离。氧化铝用稀硝酸或稀盐酸处理,可使氧化铝颗粒表面带有硝酸根或氯离子,制成酸性氧化铝,酸性氧化铝具有离于交换剂的性质。中性氧化铝使用最多,柱层析用的中性氧化铝粒度应在100~200目之间。粒度大,分离效果差:小则洗脱速度慢,纵向扩散。中性氧化铝的活化与硅胶差不多。中性氧化铝柱的洗脱溶剂溶剂组成很多,从非极性到极性的顺序洗脱。 (3)聚酰胺:尼龙与酰胺类化合物形成的高分子聚合物,分子上有很多酰胺基,可与酚类、酸类、醌类、硝基化合物形成氢键。适用于酚性化合物如黄酮的分离。一般聚酰胺不需活化或预处理就可以装柱。提取液或提取物上柱后,用水洗杂质,用不同比例的醇的水溶液从低浓度到高浓度依次洗脱。 (4)大孔树脂:具有立体结构的多孔性聚合物,有巨大的比表面积,通过范德华力吸附有机物。以前的大孔树脂多用汽油等作致孔剂,需要用丙酮加热回流来预处理,现在可购买纯度较高的,使用前只需用甲醇、或乙醇洗涤至洗出液滴在水中不产生白色即可。再生树脂的方法是,用2%~5%的盐酸浸泡洗涤,用水洗到中性,然后用5%氢氧化钠浸泡洗涤,用水洗到中性即可。树脂含水量70%左右,如不慎树脂干了,可用乙醇或丙酮浸渍处理,然后再用。按实际情况,酸、碱浓度可增减。大孔树脂类型很多,最多用的是D101。多用于皂苷、黄酮、内酯、萜类和色素的分离。提取液或提取物上柱后,用水洗杂质,用不同比例的醇的水溶液从低浓度到高浓度依次洗脱。 (5)活性炭:一般需要先用稀盐酸洗涤,其次用乙醇洗,再以水洗净,于80℃干燥后再用。上柱用的活性炭,最好选用颗粒活注炭,若为活性炭细粉,则需加入适量硅藻土作为助滤剂一并装柱,以免流速太慢。活性炭主要且于分离水溶性成分。活性炭对芳香族化合物的吸附力大于脂肪族化合物,对大分子化合物的吸附力大于小分子化合物。可将水溶性芳香族物质与脂肪族物质分开,单糖与多糖分开,氨基酸与多肽分开。提取液或提取物上柱后,用水洗杂质,用不同比例的醇的水溶液从低浓度到高浓度依次洗脱。 (6)硅藻土:采用不同pH缓冲液的硅藻土分配色谱柱,可分离生物碱、酸性生物碱和中性物质。提取液或提取物上柱后,用乙醚或氯仿洗下色素、脂肪、中性杂志,然后用胺性醇洗下生物碱。 十八烷基硅烷基硅烷碱合硅胶、其它类型键合硅胶、DEAE纤维素、其他纤维素、离子交换树脂应用的较少,这里略去。







 我要推广仪器
我要推广仪器
 下载APP
下载APP