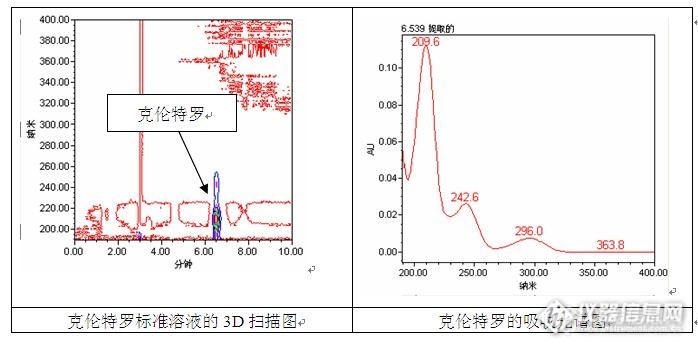
[align=center]固相萃取-高效液相色谱法测定蜂蜜中克伦特罗残留量及条件优化[/align][align=left]1、前言 克伦特罗(Clenbuterol)既不是兽药,也不属于添加剂,而是一种激素类物质,俗称“瘦肉精”,该物质能促进动物体内脂肪分解代谢,增加蛋白质合成,提高瘦肉率,然而人体食用高残留量的内脏组织或者累计摄入量超过一定值时便可能引发食物中毒事件。1997 年我国明令禁止在畜牧行业生产、销售和使用克伦特罗,但是非法使用克伦特罗的事件仍时有发生。 目前,现行有效的标准中测定克伦特罗的方法有酶联免疫法、胶体金免疫层析法、液相色谱法和质谱法,针对不同的样品基质和实验室条件可以选择合适的测定方法。国家标准GB/T 22944-2008中采用液相色谱-串联质谱法测定蜂蜜中克伦特罗的残留量,但是实验室没有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],关于高效液相色谱法测定蜂蜜中克伦特罗的文献也不多,于是尝试建立固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法并对测试条件进行优化。2、实验方法2.1 仪器和试剂 Waters e2695液相色谱仪(主要包括2998光电二极管矩阵检测器,柱温箱,自动进样器,自动脱气四元梯度泵等);微型漩涡混合仪;12位固相萃取真空装置;12位干浴氮吹仪; Millpore超纯水系统。 甲醇中盐酸克伦特罗标准溶液(250μg/mL,坛墨质检);甲醇、乙酸乙酯和乙酸为色谱纯;乙酸钠、乙酸铵、磷酸二氢钠、氨水和磷酸为分析纯;实验用水为Millpore超纯水系统制得,18MΩ• cm,25 ℃。2.2色谱条件 色谱柱:CNW Athena C18-WP(250mm×4.6mm ,5μm) 流动相:甲醇:磷酸二氢钠(0.02mol/L)=40:60 流速:1.0mL/min 进样体积:20μL 柱温:30℃ 检测波长:210nm3 结果与讨论3.1 检测波长的确定 采用Waters 2998二级管阵列检测器的3D扫描功能,在波长190~400nm范围内对高浓度的克伦特罗标准工作溶液进行测定,并在克伦特罗出峰位置提取光谱图,见下图,从图中可以看出,克伦特罗的最大吸收波长在210nm附近,因此选择210nm作为检测波长,此时克伦特罗的响应值最大。[/align][align=center][img=,690,345]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301354_01_1669358_3.jpg[/img][/align][align=left]3.2 流动相的选择3.2.1甲醇-水为流动相3.2.1.1 流动相pH值对测定结果的影响 首先参考GB/T 5009.192-2003第二法中的色谱条件,以甲醇-水为流动相进行测试,然而结果并不理想,克伦特罗标准溶液在此流动相下并未出峰,几番尝试后决定更换流动相。看到几个采用质谱测定的标准方法均在流动相中加入了酸,于是仍然以甲醇-水为流动相,往水中加入不同体积的磷酸溶液,考察pH值对克伦特罗测定结果的影响(甲醇:水=30:70),测定结果见下图。[/align][align=center][img=,583,845]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301356_01_1669358_3.jpg[/img][/align][align=left] 从上述测定结果中可以发现,未调pH时色谱图中并没有明显的色谱峰,流动相中加入磷酸后克伦特罗在8min左右出峰;随着磷酸加入量的增加,其出峰时间提前并且峰高与峰面积也随着增加;当进一步提高流动相中磷酸含量时,克伦特罗的出峰时间逐渐延长,但是峰面积保持不变。这可能是由于克伦特罗呈弱酸性,在水溶液中以离子形态存在,降低了在C18色谱柱上的保留行为,而磷酸的加入能够抑制克伦特罗的电离,使其以分子的形态存在,增加克伦特罗在C18色谱柱上的保留,并改善峰形。当pH=3.5时,克伦特罗呈部分解离的状态,因此虽然能够出峰,但是峰面积偏小,而当pH=3.0时,克伦特罗则全部以分子形态存在,峰面积保持不变。随着磷酸加入量的增加,克伦特罗的出峰时间延长,峰展宽变大,峰高变小,方法的灵敏度也随着降低。3.2.1.2 流动相比例对测定结果的影响 流动相中有机相比例对高效液相色谱的分离行为有很大影响,调节流动相比例可以改善待测组分的峰形、出峰时间以及与杂质组分的分离度,因此实验中考察了有机相比例对克伦特罗测定结果的影响(pH=2.8),测定结果见下图。从图中可以看出,提高流动相中甲醇含量对克伦特罗的出峰时间有很大的影响,当流动相中甲醇含量为20%时,克伦特罗的出峰时间为17min左右,而当流动相中甲醇含量为45%时,克伦特罗的出峰时间提前到3min左右,而且与溶剂峰重叠,不利于定性和定量分析。[/align][align=center][img=,578,826]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301359_02_1669358_3.jpg[/img][/align][align=left]3.2.1.3 方法重复性测试 根据上述测试结果,初步决定采用甲醇:水(pH=2.8)=30:70的流动相条件进行测试,然而在测试过程中发现,此流动相条件下克伦特罗的保留时间不稳定,随着进样次数的增加,保留时间不断前移,6针进样后克伦特罗保留时间的相对标准偏差(RSD)值为1.3%。产生保留时间漂移的原因可能有两种(1)色谱柱的性能下降,流动相中加了磷酸,色谱柱平衡所需的时间比较长(这是一根服役了很久的色谱柱);(2)此流动相条件的缓冲能力弱,在线混合以及样品的加入导致流动相的pH值发生变化,从而保留时间不稳定。于是修改流动相条件,pH值保持不变,将流动相中甲醇的比例由30%提高至40%,重新考察方法的重复性,实验结果见下图。实验表明,甲醇:水(pH=2.8)=40:60时方法具有较好的重复性,连续6针进样,克伦特罗保留时间的RSD值为0.1%。[/align][align=center][img=,589,419]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301401_01_1669358_3.jpg[/img][/align][align=left]3.2.2甲醇-磷酸盐为流动相 通过上述实验,初步确立了以甲醇-水为流动相测定克伦特罗的高效液相色谱条件,并对部分实验参数进行了优化,然而上述实验结果均是针对克伦特罗标准工作溶液进行的测定,样品成分单一,没有杂质干扰,因此不用考虑杂质与克伦特罗之间的分离度。色谱条件为甲醇:水(pH=2.8)=40:60时虽然解决了方法重复性的问题,但是从色谱图中可以看到,此时克伦特罗的出峰时间较早,仅需3.5min左右就能出峰,而且出峰时间早于溶剂峰,在实际样品分析中很容易受到杂质峰的影响,此方法是否适合测定蜂蜜中的克伦特罗残留量还需进一步验证。 在对蜂蜜样品测定验证之前,先尝试寻找是否有更合适的流动相。以甲醇-水为流动相进行测试时,磷酸的加入对克伦特罗的出峰影响较大,于是尝试改用甲醇-磷酸盐为流动相进行下一步测试。从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,磷酸盐选用0.02mol/L的磷酸二氢钠,通过磷酸调节流动相的pH值,实验结果见下图。实验表明,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间也有很大的影响,pH值的影响较小,因此后续的实验中直接采用甲醇-磷酸二氢钠(0.02mol/L)为流动相,未对流动相的pH值进行调节。当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,连续6针进样,克伦特罗保留时间的RSD值为0.1%,测试方法具有较好的重复性,同时,克伦特罗的出峰时间远离溶剂峰,可以减小样品中杂质组分对待测物质测定的干扰。[/align][align=center][img=,597,550]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301403_01_1669358_3.jpg[/img][/align][align=left]3.2.3甲醇-乙酸盐为流动相 在查阅文献的过程中发现,也有老师采用乙酸盐缓冲溶液对克伦特罗进行测定,于是决定试一下效果。同样,从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,乙酸盐选用0.02mol/L的乙酸铵,通过乙酸调节流动相的pH值,实验结果见下图。实验表明,采用甲醇-乙酸铵(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间有很大的影响,pH值的影响较小,方法的重复性较好,但是此时基线噪音较大,峰高变小,影响方法的检出限。[/align][align=center][img=,581,588]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301405_01_1669358_3.jpg[/img][/align][align=left] 通过上述实验,分别以甲醇-水、甲醇-磷酸盐和甲醇-乙酸盐为流动相,建立了高效液相色谱法测定克伦特罗的方法,并对部分实验条件进行了优化,在实验中发现了各自方法的优缺点,哪种方法更适合蜂蜜中克伦特罗的测定,还需用蜂蜜样品进行验证。3.3 样品前处理 蜂蜜中通常不含有克伦特罗,即使有,其残留值也很小,而且蜂蜜为半固态粘稠样品,无法直接进样测定,因此需要对蜂蜜样品进行前处理。前处理过程主要包含提取、富集、净化和浓缩4个步骤,此次实验蜂蜜中克伦特罗的提取方法参考GB/T 22944-2008:称取约2g蜂蜜样品于50mL离心管中,加入20mL乙酸钠缓冲溶液(0.2mol/L,pH=5.0),漩涡混匀,蜂蜜完全溶解后备用。 样品中克伦特罗残留量富集、净化的方法有很多种,本次实验分别参照标准GB/T5009.192-2003、SN/T1924-2007、SN/T1924-2011以及GB/T22944-2008,采用CNWBONDWCX(500mg,6mL)、CNWBOND SCX(500mg,6mL)、CNW Poly-Sery MCX(60mg,3mL)和CNW Poly-SeryHLB(500mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,其中SN/T1924-2007标准已作废,但是标准中所涉及的富集净化方法也曾出现在上海安谱实验科技股份有限公司(以下简称上海安谱)的技术应用文章中,因此对此方法也做了尝试。同时,采用Oasis MCX(60mg,3mL)和OasisHLB(200mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,简单比较了不同品牌固相萃取柱在净化效果、回收率等性能方面的差异。[/align][align=center][img=,578,358]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301407_01_1669358_3.jpg[/img][/align][align=left] 固相萃取柱所用的填料不同,活化和净化方法也有所区别,具体实验方法如下: (1)WCX固相萃取柱 依次用5mL乙醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL 乙醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-乙醇溶液(体积比2:98)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (2)SCX固相萃取柱 依次用5mL甲醇、5mL水和5mL0.03mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (3)MCX固相萃取柱 依次用3mL甲醇、3mL水和3mL 0.1mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用3mL 0.1mol/L盐酸溶液、3mL水和3mL50%甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用5mL氨水-甲醇-乙酸乙酯溶液(体积比5:45:50)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (4)HLB固相萃取柱 依次用5mL甲醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL流动相溶解残渣,过0.22μm滤膜后,进样测定。[/align][align=center][img=,578,190]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301408_01_1669358_3.jpg[/img][/align][align=left]3.4 蜂蜜样品的测定3.4.1甲醇-水为流动相 以甲醇-水(pH=2.8)为流动相,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响。 当甲醇:水(pH=2.8)=40:60时,测定结果见下图。从图中可以看出,蜂蜜样品提取后直接进样测定色谱图中,在克伦特罗保留时间处有一个很大的杂质峰,虽然该杂质峰经HLB固相萃取柱净化后消失,但是实际检测时该杂质峰对克伦特罗仍然存在隐患,如果净化不干净,就可能出现假阳性的结果,同时,在该流动相条件下,样品在2~6min内有许多杂质峰,影响克伦特罗与杂质组分之间的分离度。对比HLB与MCX固相萃取柱净化后测定的色谱图,在此色谱条件下,HLB的净化效果要优于MCX固相萃取柱,蜂蜜样品经HLB固相萃取柱净化后杂质峰明显减少,MCX固相萃取柱净化后仍有杂质组分会对克伦特罗的测定产生干扰。[/align][align=center][img=,581,863]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301411_01_1669358_3.jpg[/img][/align][align=left] 改变流动相比例,测定蜂蜜加标样品经MCX固相萃取柱净化后的样品,改善克伦特罗与杂质间的分离度,实验结果见下图。从图中可以看出,当甲醇:水(pH2.8)=30:70时克伦特罗与杂质组分的分离度较差,不能实现基线分离;当甲醇:水(pH2.8)=20:80时,克伦特罗与杂质组分虽然能实现基线分离,而且附近没有杂峰干扰,但是此时克伦特罗的峰高变小,灵敏度变低,不利于低浓度克伦特罗残留量的检查。[/align][align=center][img=,584,295]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301413_02_1669358_3.jpg[/img][/align][align=left]3.4.2甲醇-乙酸铵为流动相 以甲醇-乙酸铵(0.02mol/L)为流动相,测定MCX固相萃取柱净化后的蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当流动相为甲醇:乙酸铵(0.02mol/L)=60:40时,克伦特罗的测定受到杂质组分的影响很大,克伦特罗出峰时基线较高,而且与杂质组分不能基线分离;当流动相为甲醇:乙酸铵(0.02mol/L)=40:60时,样品中克伦特罗的灵敏度明显降低。[/align][align=center][img=,586,354]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301414_01_1669358_3.jpg[/img][/align][align=left]3.4.3甲醇-磷酸二氢钠为流动相 以甲醇-磷酸二氢钠(0.02mol/L)为流动相,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,克伦特罗与样品中的杂质实现基线分离,其保留时间附近的干扰组分较少,灵敏度能够满足残留量检测的要求。同时,在此色谱条件下,蜂蜜加标样品只需经过简单的提取便能直接测定,给高残留值蜂蜜样品的测定提供了便捷的方法(直接提取进样时的加标量远高于采用固相萃取柱净化时的加标量)。结合上述实验,最终本实验选择采用甲醇-磷酸二氢钠(0.02mol/L)为流动相对蜂蜜中克伦特罗残留量进行测定。[/align][align=center][img=,578,594]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301416_01_1669358_3.jpg[/img][/align][align=center][img=,580,634]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301417_01_1669358_3.jpg[/img][/align][align=left] 找到合适的流动相后,在该色谱条件下对固相萃取柱的净化效果进行考察。从上图可以看出,乙酸钠缓冲溶液提取后直接进样的色谱图中虽然也能检出克伦特罗,但是其加标量较大(约为固相萃取柱方法的20倍,该实验主要是为了考察杂质对待测物质的干扰),实际样品中克伦特罗的残留值远小于此次的加标量,因此实际样品测定时需要采用固相萃取柱对克伦特罗残留量进行富集、净化和浓缩。对比不同填料固相萃取柱对蜂蜜加标样品净化后测定的色谱图可以发现,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,CNWBOND SCX固相萃取柱净化后杂质组分的响应值依然很大,SN/T 1924-2007中采用了C18和SCX固相萃取柱串联的方法进行净化,而本实验仅采用了SCX固相萃取柱进行净化,这可能是导致杂质去除不完全的原因之一;采用其他几种固相萃取柱净化后,色谱图中杂质峰的响应值明显降低,其中采用WCX固相萃取柱净化后的色谱图中杂质少,基线比较平整。对比相同填料不同品牌固相萃取柱净化后的色谱图可以发现,两种品牌的固相萃取柱对杂质去除能力难分伯仲。3.5 标准曲线的绘制 准确吸取1.0mL盐酸克伦特罗标准溶液于25mL容量瓶中,用水稀释成浓度为10.0μg/mL的标准储备溶液。吸取适量体积克伦特罗标准储备溶液,用水稀释配成相应浓度的标准工作溶液,并按上述选定的色谱条件进行测定。以样品峰面积Y(mV)对质量浓度X(μg/mL)作图,得到线性回归方程Y=146893X+311,相关系数R2=0.9991,结果表明:克伦特罗含量在0.10~1.00μg/mL之间时,该方法呈现良好的线性关系。标准曲线见下图。[/align][align=center][img=,581,408]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301418_01_1669358_3.jpg[/img][/align][align=left]3.6 回收率的测定 称取约2g蜂蜜样品,共12份,往每份样品中加入0.50mL浓度为1.0μg/mL的克伦特罗标准工作溶液,样品经乙酸钠缓冲溶液提取后,分别采用上述6种固相萃取柱对样品进行净化,每种固相萃取柱做2平行,并以甲醇-磷酸二氢钠(0.02mol/L)为流动相进行测定,考察固相萃取柱的回收率,实验结果见下表。测定结果表明,6种固相萃取柱均具有较好的回收率,回收率大于85%,其中MCX和HLB固相萃取柱的回收率明显高于其他两种填料的固相萃取柱;实验中所使用的相同填料不同品牌的固相萃取柱回收率结果相近。[/align][align=center][img=,690,126]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301420_02_1669358_3.jpg[/img][/align][align=left]4、小结 (1)本实验建立了固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法,分别以甲醇-水、甲醇-乙酸盐和甲醇-磷酸盐为流动相,考察了流动相对测定结果的影响。实验结果表明,以甲醇-水为流动相时,流动相对pH值的缓冲能力较弱,克伦特罗的保留时间发生漂移;以甲醇-乙酸盐为流动相时,基线噪音较大,方法灵敏度低;以甲醇-磷酸盐为流动相,方法重复性好,灵敏度较高。 (2)实验中参考不同的标准方法对蜂蜜样品进行前处理,考察了前处理方法对样品净化和回收率影响,同时比较了固相萃取柱性能和品牌对蜂蜜中克伦特罗残留量测定的影响。实验结果表明,固相萃取柱性能对蜂蜜中克伦特罗残留量的测定结果有很大的影响,4种不同填料的固相萃取柱中,MCX和HLB固相萃取柱的回收率较高,WCX和SCX固相萃取柱的回收率略低。 (3)实验中考察了WCX和SCX固相萃取柱的回收率较低的原因:WCX固相萃取柱在乙醇淋洗的过程中会有部分克伦特罗被淋洗下来,从而回收率降低;采用SCX固相萃取柱净化时,洗脱液的碱性需要足够强,否则洗脱不完全,但是提高洗脱液的碱性后,杂质也会一同被洗脱下来。 (4)通过此次实验可以发现,流动相条件和固相萃取柱的选择对样品测定具有很大的影响,不同的流动相其洗脱能力不一样,截止波长也会有差别,实验中以甲醇-乙酸铵为流动相时基线噪音明显大于其中两种流动相;同样是MCX净化后的样品,以甲醇-磷酸盐为流动相时测定得到的样品色谱图中杂质要少于其他两种流动相。 (5)液相色谱虽然对克伦特罗具有较高的响应,但是与质谱相比,质谱具有更高的响应,因此对于残留量很低的样品,还是需要用质谱进行验证,液相方法可以作为前期的筛查手段,[/align]
固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法-串联质谱法测定蜂蜜中甲硝唑[color=black] [/color][font=宋体][color=black]甲硝唑(MNZ)属于硝基咪唑类广谱抗生素,广泛用于预防和治疗组织滴虫病、球虫病等疾病,甲硝唑因疗效明显,价格低廉,被蜂农广泛使用,造成了甲硝唑药物在蜂蜜中残留[/color][/font][font=宋体][sup][size=13px][1,2][/size][/sup][/font][font=宋体][color=black],研究发现甲硝唑对人体具有潜在的致癌和致畸作用[/color][/font][font=宋体][sup][size=13px][3,4][/size][/sup][/font][font=宋体][color=black]。1998年欧盟禁止甲硝唑使用于食品动物,2002年美国食品与药物监督管理局禁止在进口动物源性食品中使用甲硝唑[/color][/font][font=宋体][sup][size=13px][5,6][/size][/sup][/font][font=宋体][color=black]。我国农业部和国家药品监督管理局2002年规定甲硝唑及其盐、酯及制剂不准以促进动物生长为目的在所有食品动物饲养过程中使用,且不得在动物源食品中检出[/color][/font][font=宋体][sup][size=13px][7,8][/size][/sup][/font][font=宋体][color=black]。目前甲硝唑的测定方法主要有[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法、高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法、[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱法和[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法[/color][/font][font=宋体][sup][size=13px][9][/size][/sup][/font][font=宋体][color=black]。其中,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法-串联质谱法因选择性强、灵敏度高、检出限低而成为测定甲硝唑的优势方法[/color][/font][font=宋体][sup][size=13px][10][/size][/sup][/font][font=宋体][color=black]。本文将蜂蜜用乙酸乙酯萃取,提取液浓缩后经 [/color][/font][color=black]MCS [/color][font=宋体][color=black]固相萃取柱快速富集净化样品的前处理方法,减少前处理的操作步骤,同时降低基质干扰,利用超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法—串联质谱法测定蜂蜜中甲硝唑的方法,内标法定量,提高了检测效率,适合大批量样品检测。 [/color][/font][color=black]1.材料与方法[/color][size=16px][color=black] [/color][/size][color=black]1.1 仪器与试剂[/color][color=black]Waters Xevo TQ-S三重四极杆质谱仪(美国Waters),配有电喷雾离子源(ESI) Heidolph Multi Reax全能型振荡器(德国海道夫) 氮吹仪(美国Organomation);高速低温离心机(湘仪) 乙腈、甲醇(色谱纯,德国Merck);甲酸(色谱纯,上海麦克林);乙酸乙酯(色谱纯,美国Fisher);氨水(分析纯,天津科密欧);盐酸(优级纯,北京化工厂);MCS固相萃取小柱(天津,艾杰尔):500ml/6ml;甲硝唑标准品、D4-甲硝唑(纯度均大于99.0%)。实验用水为超纯水(电阻率为18.2mΩ.厘米)。[/color][color=black]1.2 样品前处理[/color][color=black]1.2.1 样品提取 称取蜂蜜5g(精确到 0.01 g)于50ml离心管中,加入100μlD4-甲硝唑内标应用液(20.0ng/ml),加水10ml,混合溶解,再加入10mL乙酸乙酯,涡旋1min,震荡提取 10min,1000rpm 离心 2min,吸取上层乙酸乙酯相 5mL 于10mL 试管,50℃氮气吹干后,加入 0.1mL 甲醇溶解,再加入 1.9mL 40mmol/L盐酸溶液,超声溶解 1min,转入 2mL 离心管,12000rpm 离心 2min,上清液待净化。[/color][color=black]1.2.2 样品净化 依次用 5mL 甲醇、5mL 水、5mL 40mmol/L 的盐酸溶液活化平衡MCS 固相萃取柱,然后转移上述上清液至 MCS 柱内,待样品过柱后,用 5mL水淋洗除杂,真空抽干柱内液体后加入 5mL 乙酸乙酯洗脱,再用 5mL 甲醇淋洗除杂,真空抽干后用 5mL 5%氨化甲醇洗脱,收集于 10mL 具塞试管内,得甲硝唑洗脱液;洗脱液在 50℃下用氮气吹干,分别先加入 0.1mL 甲醇超声溶解残留物,再加入0.9mL 10%甲醇/水溶液混匀,过 0.22μm 滤膜后待 [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 分析。[/color][color=black]1.3 仪器条件[/color][color=black]1.3.1 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]条件 色谱柱:Waters ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm),流动相A为0.05% 氨水溶液,B为乙腈,流速为 0.3 mL/min,柱温:40 ℃,进样量 5.0 μl。[/color][color=black]1.3.2 质谱条件 电喷雾离子源:ESI;质谱多重反应监测方式:MRM;正离子模式(ESI+);毛细管电压:0.5 kV;离子源温度150 ℃;脱溶剂气温度400 ℃;脱溶剂气流量800 L/h。其它质谱参数见表1。[/color][align=center][color=black]表1 [/color]甲硝唑的质谱参数与保留时间[/align][table][tr][td][align=center]化合物名称[/align][/td][td][align=center]母离子[/align][/td][td][align=center]子离子[/align][/td][td][align=center]碰撞能量(eV)[/align][/td][td][align=center]锥孔电压(V)[/align][/td][td][align=center]保留时间(min)[/align][/td][/tr][tr][td][align=center]甲硝唑[/align][/td][td][align=center]172.2[/align][/td][td][align=center]128.1*[/align][align=center]82.1[/align][/td][td][align=center]18[/align][align=center]20[/align][/td][td][align=center]54[/align][/td][td][align=center]1.40[/align][/td][/tr][tr][td][align=center]D4-甲硝唑[/align][/td][td][align=center]176.2[/align][/td][td][align=center]128.1*[/align][align=center]49.0[/align][/td][td][align=center]22[/align][align=center]22[/align][/td][td][align=center]2[/align][/td][td][align=center]1.39[/align][/td][/tr][/table]注:*为定量离子[color=black]结果与讨论[/color][color=black] 前处理方法优化 针对蜂蜜样品和目标物的性质,比较了3种不同的前处理方式,包括:(1)采用水直接溶解蜂蜜,再将蜂蜜水溶液进行固相萃取净化;(2)加水溶解蜂蜜后,加入乙酸乙酯萃取目标物,取乙酸乙酯层并将溶剂吹干后加入超纯水溶解残渣,再进行固相萃取净化;(3)采用pH=8.8的磷酸缓冲液溶解蜂蜜,再将样品溶液进行固相萃取净化.通过加标回收实验比较回收率表明,本实验采用方法(2)的回收率明显高于其他2种方式,故对蜂蜜试样采用方法(2)前处理方式。[/color][color=black] 基质效应的影响 基质和干扰组分的存在影响待测物的离子化效率,从而影响定量结果的准确性,常表现为基质增强或基质抑制效应[/color][sup][size=13px][11][/size][/sup][color=black]。分别采用空白蜂蜜,按照实验方法提取与净化后的定容液和初始流动相作为标准溶液的稀释溶剂,通过测定标准溶液的峰面积的比值考察基质效应的强弱。结果表明:两者的峰面积比值为0.757,即蜂蜜基质对甲硝唑的测定具有一定的抑制效应,本实验选择同位素内标法定量,从而有效地降低样品的基质效应的对测定结果的影响。[/color][color=black]2.3 质谱条件的优化 将甲硝唑标准工作液注入质谱,启用质谱智能方法开发程序,优化碰撞能量,碰撞池电压等参数,进一步优化其他质谱参数使灵敏度和离子化效率达到最优时保存为质谱方法。离子对、碰撞能量、锥孔电压、电离方式见表1。[/color][color=black]2.4 方法的线性关系和检出限 以甲硝唑与相应同位素内标的色谱峰面积比(y)为纵坐标,以甲硝唑的质量浓度(x)为横坐标,绘制工作曲线,线性回归方程为Y=1.004X+0.1243,相关系数r:0.9996,线性关系良好。以信噪比S/N=3时对应的浓度为方法检出限为0.05[/color]μg/kg[color=black],S/N=10时对应的浓度为方法定量限为0.15[/color]μg/kg。标准工作曲线见图1。[align=center][color=black]图1 甲硝唑工作曲线[/color][/align][color=black]2.5 方法的精密度和回收率 [/color]以5g空白蜂蜜样品作为本底,分别加入高、中、低3种不同浓度标准应用液,得到浓度为1[color=black]μg/kg[/color]、5μg/kg、20μg/kg的加标样品,充分混匀后按样品处理方法进行处理,平行测定6次,计算其加标回收率和相对标准偏差(RSD),加标回收率分别为87%~96.3%,RSD在2.23~6.17%之间,结果表明,此方法具有良好的准确性和精密度。[color=black]2.6 样品检测[/color][font=calibri][size=13px] [/size][/font][color=black]采用本方法对市售30份不同蜂蜜样品进行检测,其中1份检出甲硝唑残留,含量是0.27 [/color]μg/kg,检出率为3.3%。3 结论本研究建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法—串联质谱法测定蜂蜜中甲硝唑的含量的方法,样品前处理采用乙酸乙酯提取,固相萃取柱富集和净化,净化效果好,提取效率高。不同蜂蜜样品基质效应使甲硝唑在质谱中存在不同程度的基质抑制效应,实际测定中蜂蜜的种类繁多,若使用外标法定量应尽量使用与待测样品基质相同的样品作基质匹配工作曲线,基质不同需要配置不同的曲线系列,大大增加了工作量。本研究采用同位素内标法定量,降低了样品的基质效应的影响,只需配置一套工作曲线,提高了工作效率。本方法快速、准确、灵敏,能够满足日常蜂蜜样品中甲硝唑残留的大批量检测。参考文献[1]梁明.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法对蜂蜜中氯霉素和甲硝唑残留的测定分析[J].中国高新科技,2019(17):72-73.[2]张晓艺,张秀尧,蔡欣欣,李瑞芬.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]联用三重四极杆质谱法同时测定蜂蜜中氯霉素、甲硝唑和林可霉素[J].预防医学,2019,31(02):212-216.[3]周贻兵,吴坤,李磊,林野,刘利亚.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中甲硝唑[J].理化检验(化学分册),2017,53(08):946-949.[4]丁燕玲,陈彤,黄婷,钟名琴,吴雯娟,罗燕.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定鸡肉中甲硝唑、二甲硝唑及其代谢物的方法研究[J].广东化工,2018,45(13):245-248+252.[5]王春民,张秋萍,吴春霞.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法检测蜂蜜中的甲硝唑含量[J].食品安全质量检测学报,2016,7(05):1813-1817.[6]章剑,李昌安,李建伟,董骏,张克才.固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法同时测定蜂蜜样品中氯霉素和甲硝唑[J].安徽预防医学杂志,2018,24(01):16-20.[7]刘伟,张楠,李兵,范赛,屠瑞莹,吴国华,薛颖,赵榕.固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-同位素稀释串联质谱法测定蜂蜜中的甲硝唑和氯霉素[J].分析科学学报,2017,33(01):145-148.[8]肖国军,蔡超海,王生,覃玲.固相萃取高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱法同时测定蜂蜜中甲硝唑、氯霉素、甲砜霉素和氟甲砜霉素残留[J].中国卫生检验杂志,2018,28(01):22-25.[9]高何刚,杜赛,王瑞,陈理.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中氯霉素和甲硝唑残留[J].预防医学,2017,29(09):969-972.[10]高何刚.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]一串联质谱法测定蜂蜜中氯霉素和甲硝唑残留[J].广东化工,2017,44(15):255-256.[11]图雅,崔建平,赵宏.同位素内标-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中氯霉素及甲硝唑[J].中国食品卫生杂志,2017,29(04):450-453.
一般有机样品超声波萃取,我们使用磨口盖子的锥形瓶做器皿,样品放进去后加试剂超声,考虑超声波过程温度升高,溶剂挥发,大家如何保证器皿的密封性问题?
蜂蜜浸水果,用正己烷萃取后,萃取层乳化严重,呈泡沫状。这情况正常吗,如何解决?
各位大侠好,我最近在用固相微萃取做蜂蜜香气,总离子流图中有三四个硅类物质的峰特别高,丰度已经达到了7次方,做空样是丰度很低,请问如何解决?萃取头用的是CAR/DVB/PDMS,谢谢啊!

蜂蜜检测方案——2015版蜂蜜检测专用色谱柱 2015版新版药典的蜂蜜检测项目中,样品处理也是很重要和关键的一环。色谱柱的选择也十分讲究。以下是蜂蜜中寡糖检测的样品处理环节方法和相关仪器设备。(红字为所涉及的仪器) 2015版蜂蜜检测专用色谱柱-蜂蜜寡糖检测专用柱,固相萃取装置样品处理蜂蜜寡糖检测专用柱净化:活化:25mL水,过柱流速1滴/秒上样:2g样品溶于10mL水后,缓慢加到蜂蜜专用柱上淋洗:25mL7%乙醇水,不收集淋洗液洗脱:10mL50%乙醇水,收集洗脱液浓缩:洗脱液于65℃水浴减压浓缩至干复溶:残渣加入30%乙醇1mL,摇匀即得注:1)整个SPE前处理过程都需要用固相萃取装置,抽取真空度以确保流速;2)旋蒸浓缩时推荐用25mL鸡心瓶,防止损失样品;可少量多次添加溶剂复溶;3)活化过程中不能将液面抽干,否则会影响小柱对寡糖的保留效果。http://ng1.17img.cn/bbsfiles/images/2016/04/201604281525_591787_3073701_3.jpghttp://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif
我最近做4种磺胺类药物(磺胺嘧啶、磺胺吡啶、磺胺甲基嘧啶、磺胺醋酰钠)和两种沙星类药物(培氟沙星、氧氟沙星)的前处理富集,样品基质暂定蜂蜜,但是做了一个月了,什么的提取不上来,试过液液分散萃取(氯仿、二氯甲烷、氯苯做萃取剂)、试过液液萃取(乙酸乙酯、二氯甲烷-乙酸乙酯、乙腈-氯仿做萃取剂)还有SPE(HLB柱子),都效果不好,六种目标物没法同时富集,最多就是一两种,而且回收率不高,头疼死了,求助啊,麻烦大家给出出主意吧,我还得靠这个毕业呢http://simg.instrument.com.cn/bbs/images/default/em09509.gif
问问大家萃取有机物时都用什么仪器?有没有比较简单快捷的仪器!
实验室做很多土壤有机物固相萃取的实验,一般是取1-2g土样,样品数比较大,想用微波固相萃取的方法,这样可以省时间,请问哪种微波萃取仪比较好?谢谢!
实验室做很多土壤有机物固相萃取的实验,一般是取1-2g土样,样品数比较大,想用微波固相萃取的方法,这样可以省时间,请问哪种微波萃取仪比较好?谢谢!
FHZHJSZISO0016 水质有机氮有机磷化合物的测定液/固萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法F-HZ-HJ-SZ-ISO-016水质—有机氮、有机磷化合物的测定—液/固萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法1 适用范围本法适用于地下水、地表水、饮用水中含量大于0.05µ g/L 的有机氮、有机磷化合物的测定。有机氮、磷化合物包括莠去津、氰乙酰肼、metazachlor、乙基对硫磷、甲基对硫磷、喷达曼萨林、丙唑嗪、另丁津、西玛津、特丁津、氟乐灵、烯菌酮等。2 原理概要水样中的化合物富集在反相RP-C18 材料上或其它吸附剂上,用溶剂洗脱,然后经过气相色谱用氮磷检测器测定。3 主要仪器和试剂3.1 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](同FHZHJSZISO015),玻璃或聚丙烯的萃取柱(内装RP-C18 或其它吸附剂),真空泵或压力装配,微升注射器,玻璃纤维滤纸,杂色玻璃器皿(同FHZHJSZISO015)。3.2 主要试剂所用试剂,包括水的纯度都要达到对[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中空白峰无明显干扰的程度。水要用纯化后的水,RP-C18 或其它吸附剂,调节剂(如甲醇、丙酮),洗脱剂(如甲醇、丙酮),中和用溶液,氮、磷化合物标准溶液,惰性气体。4 过程简述4.1 采样参照ISO 5667-1 和ISO 5667-2。用清洁的玻璃瓶采集水样,用棕色玻璃瓶保存,测试样品的pH 值,在采样后立即加入适量的中和溶液调节pH 值至6~9。4.2 样品制备4.2.1 活化用溶剂洗萃取柱或玻璃柱中RP-C18 材料,溶剂体积为其体积的5 倍。再用其体积5 倍的水来洗,用湿的担体来富集。4.2.2 富集测定水的体积,使水样以小于1000mL/h 的流速流过担体。富集之后用惰性气体干燥圆筒,用溶剂洗脱RP-C18,将洗脱液和溶剂放入小的刻度瓶中,用溶剂加到刻度。4.3 测试用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行测试,需用纯水做空白实验。5 准确度及精密度多个实验室的液/固萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法用于饮用水和原水的测试数据,重现性标准偏差0.0157~0.1368µ g/L,重现性变异系数20.6%~46.3%,重复性标准偏差0.0044~0.0264µ g/L,重复性变异系数7.2%~12.6%。6 来源国际标准化组织,ISO 10695:2000(E)
FHZHJSZISO0015 水质有机氮有机磷化合物的测定液/液萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法F-HZ-HJ-SZ-ISO-015水质—有机氮有机磷化合物的测定—液/液萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法1 适用范围本法适用于饮用水、地下水、地表水、废水中悬浮颗粒物含量小于0.05g/L 时有机氮、有机磷化合物的测定。有机氮、磷化合物包括莠去津、氰乙酰肼、metazachlor、乙基对硫磷、甲基对硫磷、喷达曼萨林、丙唑嗪、另丁津、西玛津、特丁津、氟乐灵、烯菌酮等。2 原理概要水样中的有机氮、磷化合物用二氯甲烷进行液/液萃取,浓缩后,萃取出的样品通过[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]色谱、利用氮、磷检测器分析。3 主要仪器和试剂3.1 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],分液漏斗,高速搅拌器,磁力搅拌棒,蒸馏装置,微升注射器,杂色玻璃器皿。3.2 主要试剂所用试剂,包括水的纯度都要达到对[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中空白峰无明显干扰的程度。水要用纯化后的水,二氯甲烷萃取剂,溶解用溶剂(如丙酮或乙酸乙酯),无水硫酸钠。中和用溶液:1mol/L 氢氧化钠溶液,1mol/L 盐酸溶液。氮、磷化合物标准溶液。4 过程简述4.1 采样参照ISO 5667-1 和ISO 5667-2。用清洁的玻璃瓶采集水样,用棕色玻璃瓶保存,测试样品的pH 值,在采样后立即加入适量的中和用酸碱溶液调节pH 值至6~9。4.2 样品制备振摇采样瓶,倒出多余的水样,加入二氯甲烷,振摇10 分钟,转移到分液漏斗中萃取,重复萃取两次,收集萃取物,使之干燥。使用蒸馏装置将干燥过的萃取物浓缩。4.3 测试用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行测试,需用纯水做空白实验。5 准确度及精密度液/液萃取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法用于废水中氮磷化合物的测定时,回收率为83%~100%。6 来源国际标准化组织,ISO 10695:2000(E)
我们实验室做蜂蜜中的磺胺药物提取,请问哪能买到固相萃取柱里面的填料啊?价格如何呢?
我公司生产镍、铜和钴,在生产环节有萃取工艺,有哪位高手可指教一下,如何测定萃取工艺有机挥发物?需要配置怎样的仪器?多谢!
我要提取蜂蜜中的磺胺类药物,因为经费有限,所以想问下 固相萃取柱可以重复利用么?可以过多个样品么?有没有再生的方法啊?
各位好。我用固相萃取做有机磷,进行加标回收,可是回收率非常低。有机磷用的甲醇溶解,用的方法是EPA-608,我也看过别人的一些萃取方法,觉得大同小异,基本步骤都差不多,用的二氯甲烷洗脱的,可回收率就是低,各位帮忙分析一下原因,或者有好的方法可以借鉴一下。另外,请问对于水样需要调节PH值么?水样中加入氯化钠对回收率有影响么?
问题描述:如何提高水中半挥发有机污染物的萃取效率? 解答:[font=宋体]水中半挥发有机污染物的萃取通常都是用固相萃取的方式。上样流速的大小又直接影响了萃取的效率。常规固相萃取柱流速都比较低,以[/font]6mL[font=宋体]固相萃取小柱为例,上样流速最大按[/font]10mL/min[font=宋体]计,[/font]1L[font=宋体]水样需要[/font]100min[font=宋体]。对于比较大的体积,固相萃取小柱填料有限,更是难以保证使用。而盘式固相萃取因为上样流速比较大,可以显著提升效率,如[/font]47mm[font=宋体]直径固相萃取膜上样流速可达[/font]100mL/min[font=宋体],[/font]1L[font=宋体]水样只需要[/font]10min[font=宋体],效率提高[/font]10[font=宋体]倍,[/font]20L[font=宋体]水样仅需[/font]3.3h[font=宋体],大大提高了检测效率。[/font]以上内容来自仪器信息网《样品前处理实战宝典》
公司刚买了个微波消解仪。但想用来做有机物(增塑剂)的萃取,不知道和索氏提取对比的效果怎样,有没那位做过这方面的对比。请告知。感谢--
GB/T5750饮用水中气相色谱法项目,很多前处理是萃取浓缩然后上气相色谱分析。例如:饮用水中邻苯二甲酸二(2-乙基己基)酯,是取500ml水加环己烷萃取浓缩至1ml;环氧氯丙烷是取100ml水样用二氯甲烷萃取浓缩至1ml。那么一个方法的萃取浓缩的最大倍数是多少?理论上如果取大量水样用有机物萃取然后浓缩至1ml,那么液液萃取方法可以做到任何检测限?
有机萃取带盖试剂管大家选择哪种?气密性等有什么要求?
为什么用二氯甲烷萃取有机氯后,还要把溶剂转换为正己烷才能用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,不转换溶剂会有什么影响吗?而用石油醚萃有机氯时,需要用硫酸磺化后才能检测?这几点我始终弄不明白,还望各位老师指教!还有萃取DDT和六六六时用哪一种萃取剂效果更好呢,谢谢![em61]
哪位老师教下我,有机汞的萃取。国标上用的是疏基棉管萃取的。现在最新的萃取用的是哪种方法啊?最好是简单、方便的。还有,现在有萃取管成品吗?
[font=宋体]链接:[/font]https://bbs.instrument.com.cn/topic/7870043问题描述:[font=宋体]水质样品有机物分析测试过程中,液[/font][font='Times New Roman','serif']-[/font][font=宋体]液萃取中萃取剂的选择原则及操作步骤是什么?[/font]解答:[font=宋体]选择合适的萃取剂主要根据待测物的性质,最根本的原则是相似相溶原理,此外也需要满足以下几点:[/font]a [font=宋体]萃取剂和溶液中的溶剂(水相)互不相溶。[/font]b [font=宋体]待测物在萃取剂和原溶液(水相)中的溶解度不同,一般在萃取剂中的溶解度远大于在水相中的溶解度。[/font]c [font=宋体]萃取剂有好的选择性,应对待测物溶解度大,对杂质和基体溶解度小。[/font]d [font=宋体]萃取剂能使萃取相和萃余相在操作条件下较快分层。[/font]e [font=宋体]沸点低,为萃取后方便蒸发溶剂,浓缩样品。[/font]f [font=宋体]纯度高,以防引进杂质。[/font]g [font=宋体]化学稳定性好,萃取剂不易水解,加热不易分解。[/font]h [font=宋体]在以上基础上尽量选择毒性小、无特殊气味的萃取剂。[/font][font=宋体]注意:原溶液通常为水相时,那么甲醇、乙醇、丙酮虽然是有机试剂,但不易作为萃取剂,因为这类萃取剂属于水溶性有机溶剂,和水互溶不能分层,达不到分离的目的。日常针对水质样品有机物分析,我们经常使用的萃取剂有正己烷、二氯甲烷、苯、甲苯、三氯甲烷、四氯化碳、乙酸乙酯等以及相应配比的混合溶剂。[/font][font=宋体]液[/font]-[font=宋体]液萃取法操作主要分为以下五个步骤:[/font]a [font=宋体]检漏并清洗瓶壁:使用分液漏斗前,要检查漏斗是否漏液并清洗瓶壁。如果出现漏液情况,会造成样品损失,严重的情况下需要重新采集样品。具体步骤为:关闭活塞,向漏斗中加入少量的农残级丙酮,看聚四氟乙烯旋塞处是否漏液。如果不漏则盖好漏斗瓶口的玻璃塞(如果漏液则立即更换旋塞直至不漏),右手握住漏斗上口的颈部,手心压紧玻璃塞,左手紧握活塞,将漏斗倒转过来,看上口处是否漏液,如果不漏则轻轻振摇丙酮溶液(如果漏液则需要更换玻璃塞至不漏),清洗瓶壁,清洗完后,左手旋转活塞轻轻放气。接着将玻璃塞旋转[/font]180[sup]o[/sup][font=宋体],再倒转漏斗检查是否漏液,如果不漏液,则可以放心使用。[/font]b [font=宋体]转移样品、加替代物和萃取剂:转移样品前一定要先关闭活塞,将样品无损转移至相应容量的分液漏斗中,并加入相匹配的替代物及萃取剂,盖紧玻璃塞。[/font]c [font=宋体]振荡:先手动振摇两次,并放气之后将分液漏斗置于振荡器上固定,振摇数分钟。[/font]d [font=宋体]静置分层:振摇完毕后将分液漏斗置于样品架上静置分层,可在此时打开顶部玻璃塞,一般静置[/font]10min[font=宋体]。[/font][font='Times New Roman','serif']e [/font][font=宋体]分液:待液体分层后,从下端放出下层液体,上端倒出上层液体,收集含待测组分相。[/font]以上内容来自仪器信息网《样品前处理实战宝典》
请教高手:1 沉积物(底泥)中有机氯农药分析时萃取一般用什么溶剂比较好?以前试过正己烷和二氯甲烷的混合液,感觉不怎么好。还有很多混合液如正己烷和丙酮,石油醚和丙酮等等,哪个效果好点?比例关系是多少?(我用的是超声萃取)2 净化时用硫酸和铜粉有什么区别?哪个较好,两个可以一起用吗?净化时应该注意什么?(之后还要过硅胶拄)
最近对植物叶中的有机酸(如草酸、丙二酸、柠檬酸、苹果酸、丁二酸和硬脂酸等等)甲酯化后,那么如何萃取出来,然后GCMS测试呢?看了很多文献都说用二氯甲烷萃取,但是觉得太烦了,有的文献用正己烷,但觉得正己烷是非极性的好像不能萃取吧,或者萃取效果很差,是这样吗?能否有一种溶剂和甲醇不溶的试剂萃取呢?急盼回答!!
想用固相微萃取技术处理废水中的有机物,有谁实际操作吗?
请教高手: 1 沉积物(底泥)中有机氯农药分析时萃取一般用什么溶剂比较好?以前试过正己烷和二氯甲烷的混合液,感觉不怎么好。还有很多混合液如正己烷和丙酮,石油醚和丙酮等等,哪个效果好点?比例关系是多少?(我用的是超声萃取) 2 净化时用硫酸和铜粉有什么区别?哪个较好,两个可以一起用吗?净化时应该注意什么?(之后还有过硅胶拄)
题目:Comparison of the performance of conventional, temperature-controlled, and ultrasound-assisted ionic-liquid-dispersive, liquid-liquid microextraction combined with high-performance liquid chromatography in analyzing pyrethroid pesticides in honey samples摘要:This research paper presents a comparative study of the performance of conventional, ultrasound-assisted (UA), and temperature-controlled (TC) ionic liquid (IL) dispersive liquid-phase microextraction (IL-DLLME). Various parameters that affect extraction efficiency, such as type and volume of extraction and disperser solvent, centrifugation time, salt addition, effect of temperature on TC-IL-DLLME, and effect of sonication time on UA-IL-DLLME, were evaluated. UA-IL-DLLME was found to provide the best extraction efficiency. Under optimized conditions, great enrichment factors (506−515) and good recoveries (101.2%−103.0%) were obtained by analyte extraction in real samples. The limit of detections (LODs) ranged from 0.21−0.38 μg/L. Good linearity was obtained in the range of 0.5−200 µg L-1 for ethofenprox and tetramethrin, and 1−200 µg L-1 for meperfluthrin and alpha-cypermethrin. Based on optimized conditions, the UA-IL-DLLME method was applied and combined with high-performance liquid chromatography with diode array detection (HPLC-DAD) to determine the presence of ethofenprox, tetramethrin, meperfluthrin, and alpha-cypermethrin in honey samples.概述:对比了传统离子液体液液分散微萃取、温控辅助液液分散微萃取和超声辅助液液分散微萃取的区别,通过优化几个影响萃取实验的参数,如:萃取剂和分散剂的种类和体积、离心时间、盐效应、控温温度和超声时间等确定萃取条件,实验结果表明超声辅助液液分散微萃取的萃取效率要高于另外两种,在优化条件下四种菊酯类农药的富集倍数达到了506-515倍,在1−200 μg/L范围内具有良好的线性关系,线性相关系数在0.9990~0.9999 之间;检出限为0.04~0.10 μg/L(S/N=3),回收率为101.2%−103.0%。本方法已应用实际蜂蜜样本的检验,具有操作简单、富集效率高、灵敏度高和绿色环保等特点,可满足液体样品中菊酯类农药残留的检测要求。
蜂蜜是颇受青睐的养生保健食品之一,然而,近年来,国内部分蜂蜜生产企业存在掺杂掺假,以假乱真,以次充好的现象,极大损害了我国天然蜂蜜的产品信誉。为此,月旭科技特推出伪劣蜂蜜检测即蜂蜜中寡糖测定解决方案,希望对我国蜂蜜产品的质量安全提供保障。1、适用范围适用于2015版《中国药典》一部蜂蜜中寡糖的检查。2、方法原理试样中的高分子糖,经活性炭-硅藻土小柱的富集、浓缩,再用薄层色谱法分离、检测。经过与麦芽五糖对照品比较,来判定蜂蜜中寡糖的存在。3、样品和试剂对照品:麦芽五糖供试品:纯正蜂蜜、掺假蜂蜜试剂:乙醇(分析纯),正丙醇(分析纯),三乙胺(分析纯),二苯胺(CAS:537-67-7),丙醇(分析纯),苯胺,磷酸(优级纯),超纯水。 4、样品前处理Welchrom®HON蜂蜜寡糖检查专用柱净化;活化:25ML水,过柱流速1滴/秒上样:2g样品溶于10ml水后,缓慢加到蜂蜜专用柱上淋洗:25ml7%乙醇水,不收集淋洗液洗脱:10ml50%乙醇水,收集洗脱液浓缩:洗脱液与65℃水浴减压浓缩至干复溶:残渣加入30%乙醇1ml,摇匀即得 薄层色谱法条件:TLC板:高效硅胶G薄层板(涂层厚度0.2-0.25mm),110℃活化1小时展开剂:正丙醇-水-三乙胺=60:30:0.7显色剂:苯胺-二苯胺-磷酸混合溶液(取二苯胺1g,苯胺1ml,另算5ml,加丙醇至50ml,混匀)注:显色剂最好现配现用注:整个SPE前处理过程都需要用固相萃取装置,抽取真空度以确保流速;旋蒸浓缩时推荐用25ml鸡心瓶,防止损失样品;可少量多次添加溶剂复溶;活化过程中不能将液面抽干,否则会影响小柱对寡糖的保留效果。 5、实验经过判定从恒温干燥箱中取出薄层板,观察TLC板的显色情况,纯蜂蜜只有Rf值0.35以上的区域有2-3个蓝色中带灰色或咖啡色的斑点,相比较麦芽五糖或会有麦芽五糖的蜂蜜确实从原点开始呈现蓝色带状样的斑点群。http://ng1.17img.cn/bbsfiles/images/2016/01/201601270952_583578_1863087_3.jpg对照品和供试品的薄层色谱图(a-麦芽五糖对照品,b-纯正蜂蜜,c/d/e/f-掺加蜂蜜)http://ng1.17img.cn/bbsfiles/images/2016/01/201601270953_583579_1863087_3.png原文链接:http://www.welchmat.com/yysjk/info_14.aspx?itemid=19582&categoryid=4资料下载:http://www.welchmat.com/qt/index_22.aspx
[color=#444444]我想测定污水中7种痕量有机污染物。但是水中的痕量有机污染物很多,有一个问题[/color][color=#444444]我现在只测7种物质,但是固相萃取后水中的物质可能很多,我怎么样才能准确定量我的目标物质?(因为我做标线时只有这7种物质,但是实际水样中的物质种类可能会很多,用标准物质做出来我可能得到了分离很好的出峰,但是用同样的方法测实际水样时的出峰可能会和其他非目标物质重合)[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP