请教:用纳氏试剂分光光度法测定废水中有机胺的氨氮含量,测量值和计算值有一定差距(偏小20%以上)。废水中的有机胺是季胺盐(四丙基溴化铵),按照国标中的原理季铵盐是否能和铵离子一样和纳氏试剂形成淡红棕色的络合物。用现行的国标规定的方法,能否有效地检测有机铵的氨氮含量?
有机涂料与塑料等好像都以三聚氰胺做为增塑剂,不知道现在饮料用的有机塑料瓶能否检测出三聚氰胺呢?
请问各位,知道怎么测定液体中有机胺和无机氨吗?无机氨可能是氨水,该怎么区分呀?
极性柱子能否测类似二异丙胺那样的强碱性胺类有机溶剂?我进样检测出峰很怪,拖尾现象很严重,基线不是很稳,为排除是样品的问题,进乙醇样正常。我用的是极性柱是岛津的WondaCAP WAX。如果测不了强碱性胺类有机溶剂,有什么办法可以测?测定后会不会对柱子伤害很大?
让全国人们关注的三聚氰胺事件愈演愈烈。三聚氰胺之所以被不法分子加入到奶制品当中,是因为三聚氰胺的含氮量非常高,达60%以上,可以提到奶制品的粗蛋白检测含氮量。那么除了三聚氰胺之外,还有什么别的有机物含氮量非常高。不法分子以后虽然不会添加三聚氰胺,那是否还会添加别的物质?!!所以希望大家把一些含氮量高的有机物都贴出来,谢谢1
问题: 请问各位大神,有机碱该怎么测啊?像乙醇胺
有机酰胺系复合物对甲醛有吸附作用吗?
我现在在做有机叔胺的非水滴定,遇到一个头疼的问题,想请教高人,先谢谢了.样品是一个含三个氮的六元环,三个氮上分别又连有含叔氨基的丙烷基,也就是一个六个氮,三个在环上,三个在支链上,都是叔胺.用0.1M高氯酸滴定 ,100ml醋酸+7ml醋酐作溶剂,出两个峰,第一个峰最大,比理论值小1/3,第二个峰小很多,和理论值接近,我想问下是该取第二个峰吗?做平行样时发现, 第一个峰很好平行,第二个峰会差比较多,想用第二个峰又不好平行,疑惑于到底该取哪个峰,跪求赐教!
如题!做饮料(如可乐)中的有机胺,怎样前处理好呢?用210nm检测,干扰太多了。
求助有关水中有机胺的测定的分析方法,能说说分析的关键和注意事项更好,多谢!
氢氧化钠中含有有机胺,如何检测二者含量啊?
最近测一个水溶性样品的三乙胺和其他有机溶剂的残留,被三乙胺搞得有点小郁闷。之前用直接进样的方法,用水做溶剂,中等极性的毛细管柱,三乙胺出了一个馒头峰,峰很宽,灵敏度很低,不成线性。听说是三乙胺和水氢键结合了。后来换成用DMSO或DMF溶,灵敏度提高很多,但样品不溶于这俩溶剂中。于是打算用顶空进样,还是用水溶,试问这样能否改善三乙胺的灵敏度问题,比直接进样好些?能摆脱水对三乙胺的干扰?
弱弱的问一句:有机溶剂残留中三乙胺的限量是多少?指导原则上找不到呢。。。顺便问下在指导原则上找不到的有机溶剂去哪儿找它们的限量??坐等回复,谢谢啦
各位老师,请教一下:[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测定有机胺,有时需要二次曲线定量,二次曲线算是呈线性吗?线性必须是直线线性吗?请各位大神赐教,感谢!!
如题,有机胺溶液中含少量氢氧化钠,如何区分测定,计划用吡啶做溶剂做非水滴定,用什么指示剂好?有无可行性?
[b][font=宋体]问题描述:如何使用液相色谱检测有机胺类化合物?使用[/font]C[sub]18[/sub][font=宋体]色谱柱还是氨基柱?加入氨水或者缓冲盐的比例是多少[/font]?[font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])根据有机胺类化合物的结构式查询并确定其性质属于中性、酸性或者碱性,一般来说中性有机胺类化合物直接使用水[/font]+[font=宋体]有机溶剂即可[/font],[font=宋体]如果柱子保留较弱也可以加点氨水,而酸性和碱性有机胺类化合物则需要使用缓冲盐。[/font][font=宋体]([/font]2[font=宋体])一般有机胺类化合物的液相检测,色谱柱可以直接使用[/font]C[sub]18[/sub][font=宋体]色谱柱,而如果使用了高[/font]pH[font=宋体]值的缓冲盐或者因为极性较强保留较弱的情况下,则需要用到[/font]HILIC[font=宋体]色谱柱。[/font][font=宋体]([/font]3[font=宋体])如果需要加氨水的情况下,氨水一般在[/font]0.05%~0.1%[font=宋体]浓度,而缓冲盐的浓度一般为[/font]0.02mol/L[font=宋体]左右。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
在做蔬菜农残时,以前有机磷混标(1mg/L)的各峰高均差不多高。现就不一样了,乙酰甲胺磷的峰几乎是原来峰的10%了,而甲拌磷比原来的倒高了,为什么?真的纳闷呀
聚丙烯酰胺不溶于那些有机溶剂?此溶剂要能够溶解氯化钠、葡萄糖、氯化铵,元明粉等物质。谢谢!
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
采血管类型对测定有机磷农药或毒鼠强或氟乙酰胺有影响吗?如肝素纳管,EDTA管,或促凝管,无抗凝管等对测定血中机磷农药或毒鼠强或氟乙酰胺结果有影响吗?
[color=blue][size=4][B]pingguwu[/B]老师在版面中发了一份“水质检测-有机磷征求意见稿”,其中有水中甲胺磷检测的操作方法,各位朋友看看内容,其前处理可行吗?数据有这么好吗?有人按照该法做过甲胺磷吗?有条件的朋友,能否验证一下此方法(主要是甲胺磷)。将结果与大家分享一下,以便于大家的交流与讨论。[/size][/color][B]下面是参考资料:[/B][url=http://www.instrument.com.cn/bbs/download.asp?ID=91639]水质检测-有机磷征求意见稿[/url]其他相关甲胺磷测定的帖子(参考):1.甲胺磷检测(1)—关于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲胺磷出峰问题的总结: http://www.instrument.com.cn/bbs/shtml/20080527/1277516/2.甲胺磷检测(2)—关于水中甲胺磷的检测方法: http://www.instrument.com.cn/bbs/shtml/20080529/1281588/3.浓缩果汁中甲胺磷的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检测: http://www.instrument.com.cn/bbs/shtml/20080522/1271738/4. 玉米中甲胺磷的GC/MS检测: http://www.instrument.com.cn/bbs/shtml/20080420/1229723/
ECD用于测敌敌畏、甲胺磷等有机磷农药可以么?
有机胺盐类化合物的核磁共振做碳谱好还是氢谱好?
我是初次来这里,不知这个问题在这里发对不对,我朋友想作有机试剂:H酸亚甲胺(亚甲胺H酸),可不知其市场前景怎么样,就让我来看看,我就找到这里了,如果大家对这有兴趣的话,也给我发信:xiaolala009@163.com欢迎大家提供信息和问题
有机磷(毒死蜱、倍硫磷、喹硫磷、马拉硫磷、杀螟硫磷、甲胺磷、甲基嘧啶磷、甲基毒死蜱、水胺硫磷、敌敌畏、乐果、乙酰甲胺磷),请问这12种有机磷农药残留可以混合在一起分析吗?我今天将它们混合配在一起,可是只出了11个峰,还有一个峰不知道与哪个峰没有分开,反正是少了一个峰。我的色谱条件:RTX1701 30m*0.25mm,0.25um,进样口:250℃ 色谱柱: 80℃(保持1min) 然后以30℃/min升至130℃,再以5℃/min升至250℃并保持8min,总运行时间44.65min,FPD检测器温度280℃
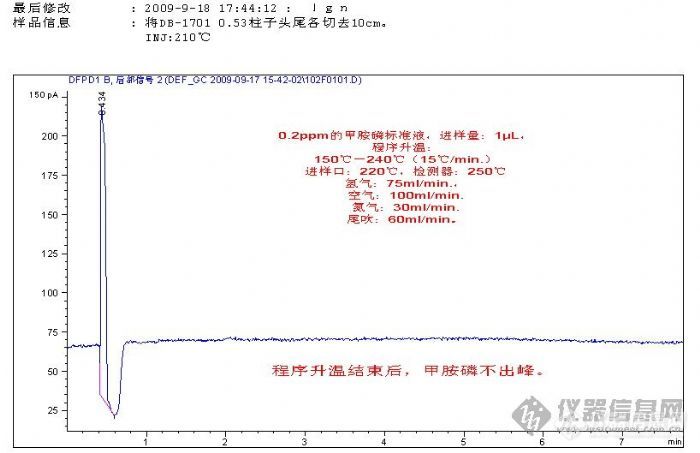
最近开展茶叶中有机磷项目的检测。购回甲胺磷、乙酰甲胺磷、毒死蜱、杀螟松4个单体标准溶液,浓度是100μL/mL.。仪器:Agilent 7890A.配双FPD检测器(DFPD),以下用谱图说明实验进程。[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909191600_171983_1620184_3.jpg[/img]
“关于水中(果汁)有机磷测定征求意见稿中的甲胺磷测定问题”对“水质检测-有机磷征求意见稿”讨论的这个帖子的已经结束,非常感谢各位版友的参与。“水质检测-有机磷征求意见稿”在原帖链接: http://www.instrument.com.cn/bbs/shtml/20080603/1287797/下面是一部分版友的意见:[B]shisheng2008:[/B][color=#00008B]1.4ppb说的是方法的检出浓度,还是i可能的。有个地方就是H2的流量不会哪么大吧!H2:Air的流量有点像FPD的。怀疑氧化乐果的回收率数据,甲胺磷回收率个人做过和他数据差不多水平,但氧化乐果回收率从来没上过90%。而且做为标准那谱图也有点太。。。。。。还不如不要放上去呢。7.3.1也是不合理的,做水样很少人用低于50mL做GC的,至少都是100mL以上。同意楼上,只要一个步骤就好。而且标准溶液进样浓度很大啊,结果会有意义?[/color] [B]pingguwu:[/B][color=#DC143C]该方法7.3.1甲胺磷的提取,既然可以提取甲胺磷,那一定可以提取其他有机磷.本方法其实还是两种样品处理方法.既然是有机磷的测定,就应该包含其他有机磷,好象7.3.2是多余的.采用液液萃取甲胺磷就必须使用大量溶剂,从某种角度,该方法的先进性是没有的,这一点制标单位也许根本没有前处理经验.[/color][B]xyzwz123:[/B][color=#00008B]看了该标准,觉得毫无新意,只是从心底里佩服标准制定者的勇气,这样的烂东西还敢拿出来,特别是哪张烂色谱图(安公司看了可能也会很生气!),真是笑死人了,一看制定者的单位都是吓死人的单位,可东西就是这么烂.下面简单说一下它的"烂"处:1.敌敌畏和甲胺磷的最低检出浓度0.0016和0.0014mg/L,应该都写成0.002mg/L2.作为国标方法应有一定的包容性,要代表国家的水平,我觉得要有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]MS方法(我做过甲胺磷和敌敌畏等,水样过滤膜后直接进样,检出限很容易达到0.1ng/L),有GC-MS方法,有GC-FPD方法,最后才是GC-NPD方法,不能限定人家只能做GC-NPD方法(更可笑的是计量认证限定大家不能做自己开发的方法)3.既然最低检出浓度如此低,为何加标回收试验的加标水平如此高,加标最低的水平约为最低检出浓度的50倍,按欧盟农残检测规定:在定量限浓度水平加标,回收率要在70-120%之间,RSD20%,这能达到吗?4.前处理比较复杂,要萃取三次,然后旋蒸,精密度如此好让人觉得很不可信!纯溶剂标准溶液连续进6针恐怕也达不如此好的精密度.不说了,国产的就是这个样子,不像EPA方法那么严密.[/color][B]wphlr:[/B][color=#DC143C]我没做过水中的,但以前做过果汁中的甲胺磷,用的是[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]MS.看了文章有几个疑问1.整个方法只有提取方法,没有净化过程,那么如果是工业废水呢?会不会有很大的干扰呢?2.乙酸乙酯能否将甲胺磷从水中完全提取出来?3.我不怀疑方法检测限,因为取样量大,达到这样的检测限是完全可以的,但是回收率真有那么好吗?平常做甲胺磷的时候从没有过如此好的回收率.4.如果有干扰,那么如何定性和定量?采用双柱,如何来判断?[/color][color=#00008B][size=4][B]如果大家还有建议和观点请多多的交流。[/B][/size][/color]
请问在什么地方可以做有机药物中三乙胺溶剂残留量?
实验室要求增加这2种农药检测,现有条件安捷伦7890A,FPD检测器,DB-1701柱,昨天做了单标测试1PPM的,结果杀扑磷峰型很差且峰面积才200多,亚胺硫磷不出峰。这个条件检测其他有机磷混标400ppb,还是很好的。求教大侠们 这2种农药是不是这个条件满足不了呢?有方法的推荐一下。
在做饮用水中有机物指标,方法中测定丙烯酰胺,前处理中为为什么4℃冰箱中溴化?,温度要求有那么严格吗,是哪一步反应需要那么低温度?







 我要推广仪器
我要推广仪器
 下载APP
下载APP