含量的高低会不会影响有关物质的高低,含量高,有关物质就会低,含量与有关物质之间的关系,有没有大佬解释一下,萌新化验员
各位兄弟姐妹: 大家平时做有关物质、含量样品检测时时怎么操作的?我们 含量测定样品称取各两份,一份进两针,一份进三针,然后五针计算RSD 有关物质只称取一份,进五针。 觉得挺麻烦的。大家都是怎么弄的?
最近在帮公司测一个药物新剂型的含量,按照公司的任务标准,除了要测主药物含量外,还要测有关物质的。任务书上是这样写的,测1%和1000%的有关物质,请问这是什么意思呢?对于测有关物质有没有统一的标准,比如说要测多大的浓度?分析多长时间?
如果制剂的含量测定和有关物质检查的条件一样,在做方法学比如线性,准确度(加入回收率)的时候,可不可以一起做,还是先做有关物质,然后做含量测定?
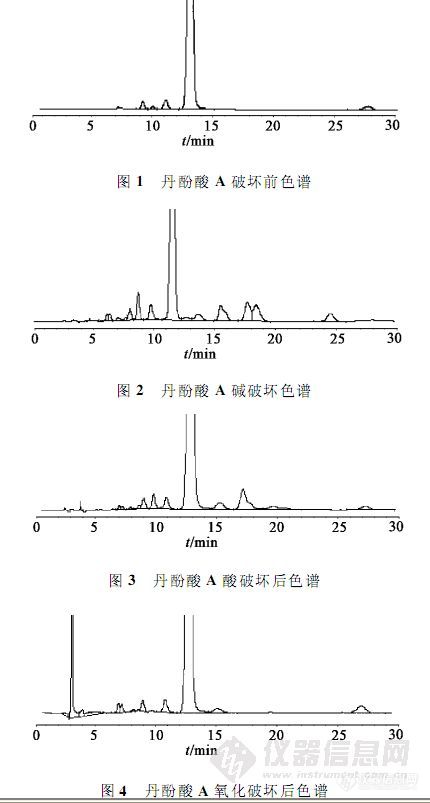
【作者】 于翠翠; 刘军锋; 车鑫; 刘珂;【机构】 烟台大学药学院; 山东靶点药物研究有限公司;【摘要】 目的:建立一种测定丹酚酸A中有关物质含量的方法。方法:用外标法测定丹酚酸C的含量,采用Diamonsil C18柱(4.6 mm×250 mm,5μm),乙腈-0.2%磷酸水溶液(30∶70)为流动相,流速1 mL.min-1,检测波长285 nm,柱温30℃,以主成分自身对照法控制其他杂质的总含量。结果:丹酚酸C的含量均1.0%,其他杂质峰面积和3.0%。结论:该方法专属性强,灵敏度高,简单准确,能有效控制丹酚酸A中有关物质的含量。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207231559_379228_2379123_3.jpg
做影响因素试验哦 有关物质有一定的变化规律 可是做出来的含量却是高高低低 不能与之保持相对一致 很困惑 这样的含量做出来还有意义么
现在有一个问题想请教大家,原先做的有关物质和含量液相条件都一样,所以做含量液相条件方法学时就不存在色谱条件确定这项了,但现在有关物质和含量液相色谱条件不一样?含量的色谱条件也点进行确定,我现在想问,含量的色谱条件也点做耐用性、专属性、检测限、溶液稳定性试验吗?
求助高效液相色谱法测盐酸氯卡色林有关物质及含量的测定[b],[color=#444444]还有手性纯度的测定方法。[/color][/b]
请问一下中国药典 05版第二部 头孢氨苄的有关物质和含量测定是怎样计算取样量的 大家能细细说一下吗本人很笨 数学学的不太好
有关物质分析方法验证的可接受标准简介摘要:本文介绍了在对有关物质检查所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。 关键词:有关物质检查 分析方法验证 可接收标准 药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至一定的浓度范围内配制6份浓度不同的供试液,分别测定该杂质峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。 3.精密度 1)重复性 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于15%。 2)中间精密度 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于20%。 4.专属性 可接受的标准为:空白对照应无干扰,该杂质峰与其它峰应能完全分离,分离度不得小于2.0。 5.检测限 杂质峰与噪音峰信号的强度比应不得小于3。 6.定量限 杂质峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±5nm、流速相对值变化±20%以及采用三根不同批号的色谱柱进行测定时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:各杂质峰的拖尾因子不得大于2.0,杂质峰与其他成分峰必须达到基线分离;各条件下的杂质含量数据(n=6)的相对标准差应不大于2.0%,杂质含量的绝对值在±0.1%以内。 8、系统适应性 配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%,保留时间的相对标准差应不大于1.0%。另外,杂质峰的拖尾因子不得大于2.0,理论塔板数应符合质量标准的规定。 9.溶液稳定性 按照分析方法分别配置对照品溶液与供试品溶液,平行测定两次主成分与杂质的含量,然后将上述溶液分别贮存在室温与冰箱冷藏室(4℃)中,在1、2、3、5和7天时分别平行测定两次主成分与杂质的含量。 可接受的标准为:主成分的含量变化的绝对值应不大于2.0%,杂质含量的绝对值在±0.1%以内,并不得出现新的大于报告限度的杂质。含量测定分析方法验证的可接受标准简介摘要:本文介绍了在对含量测定所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。关键词:含量测定 分析方法验证 可接收标准在进行质量研究的过程中,一项重要的工作就是要对质量标准中所涉及到的分析方法进行方法学验证,以保证所用的分析方法确实能够用于在研药品的质量控制。为规范对各种分析方法的验证要求,我国已于2005年颁布了分析方法验证的指导原则。该指导原则对需要验证的分析方法及验证的具体指标做了比较详细的阐述。但是文中未涉及各具体指标在验证时的可接受标准,国际上已颁布的指导原则中也未发现相关的要求。另一方面,大多数药品研发单位在进行质量研究时,已逐步认识到分析方法验证的必要性与重要性,大都也在按照指导原则的要求进行分析方法验证,但验证完后却因没有一个明确的可接受标准,而难以判断该分析方法是否符合要求。本文结合国外一些大型药品研发企业在此方面的要求,提出了在对含量测定方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。1.准确度该指标主要是通过回收率来反映。验证时一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。2.线性线性一般通过线性回归方程的形式来表示。具体的验证方法为:在80%至120%的浓度范围内配制6份浓度不同的供试液,分别测定其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。3.精密度1)重复性配制6份相同浓度的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。2)中间精密度配制6份相同浓度的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于2.0%。4.专属性可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。5.检测限主峰与噪音峰信号的强度比应不得小于3。6.定量限主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。7.耐用性分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、流速相对值变化±20%时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。8、系统适应性配制6份相同浓度的供试品溶液进行分析,主峰峰面积的相对标准差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。本文为转帖!
急求牛磺胆酸钠原料药的有关物质或者含量测定的方法?哪位老师有,能否借鉴一下
液相在做含量或有关物质分析时,都要走空白的,如果先单针走空白,再跑含量或有关物质的序列可以吗?还是必须和含量测定在同一个序列跑呢?

样品制备制备方法:【有关物质】取本品适量,加流动相A溶解并稀释至每1ml中含1.0mg的溶液,滤过,取续滤液作为供试品溶液;精密量取1ml,置100ml量瓶中,用流动相A稀释至刻度,摇匀,作为对照溶液。取7-氨基去乙酰氧基头孢烷酸对照品和α-苯苷氨酸对照品各约10mg,精密称定,置同一100ml 量瓶中,加pH7.0磷酸盐缓冲液约20mL超声使溶解,再用流动相A稀释至刻度,摇匀。精密量取2 ml,置20 ml量瓶中,用流动相A稀释至刻度,摇匀,作为杂质对照品溶液。【含量测定】系统适应性试验: 取供试品溶液适量,在80℃水浴中加热60min,取20μl,测定,头孢氨苄峰与相邻杂质峰的分离度应符合要求。含量测定法:取装量差异项下的内容物,混合均匀,精密称取适量(相当于头孢氨苄0.1g),置100 ml量瓶中,加流动相适量,充分振摇,使头孢氨苄溶解,再用流动稀释至刻度,摇匀,滤过,精密量取续滤液10mL,置50mL量瓶中,用流动相稀释至刻度,摇匀,取20μl,注入液相色谱仪。分析条件【有关物质】色谱柱:Spursil C18,150×4.6 mm,5um,Cat#:(82001)流动相:流动相A为0.2mol/L磷酸二氢钠溶液(用氢氧化钠调pH至5),流动相B为甲醇洗脱方式线性梯度流速:1mL/min柱温:30 ℃检测器:UV 220nm进样量:20 μL【含量测定】色谱柱:Spursil C18,150×4.6 mm,5um[/fon
麻烦各位老师解一下惑:有关物质检测,是否需要同含量检测一样进行平行样检测,若需要,法规上是否有依据?谢谢!
马上准备要做稳定性试验了,有一些问题向大家请教下,烦请大家不吝赐教,先谢过了!1、做影响因素长期、加速试验时,每次取样做含量测定和有关物质检查时,是否都要重新做标曲?2、 用外标法定量时,对照品溶液可否在冰箱存放,隔月再用呢?对照品溶液最多可以用多长时间呢?3、 加速试验、长期试验做满6个月后,如何做数据的统计分析呢?有效期又该如何算呢?请做过的老师指点迷津。
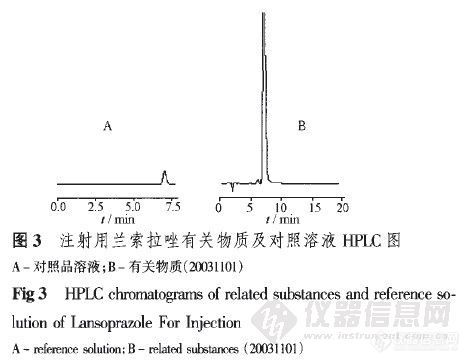
HPLC测定注射用兰索拉唑中兰索拉唑的含量和有关物质 杨艳,谢俊霞,李冰,孙英华,方金玲,刘艳,何仲贵”(沈阳药科大学药学院,沈阳110016)摘要:目的采用高效液相色谱法建立注射用兰索拉唑有关物质检查及其含量测定方法。方法Diamonsil—c18柱(4.6 l_mm×200 mm,5um),以甲醇.水一三乙胺.磷酸(640:360:5:1.5,pH为7.3)为流动相,检测波长为284Ⅲn。结果制剂中辅料对主药测定无干扰,兰索拉唑与有关物质完全分离。在10.0~400.0 mg·L一内峰面积与浓度呈良好的线性关系。精密度(RsD=0.13%)良好。平均回收率为99.6%。结论 本方法简便,迅速,准确,专属性强。关键词:高效液相色谱法;注射用兰索拉唑;含量测定;有关物质http://ng1.17img.cn/bbsfiles/images/2012/07/201207241917_379474_2355529_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207241917_379475_2355529_3.jpg
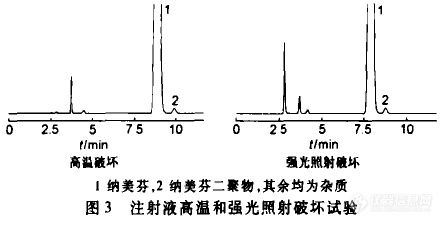
作者:钟武; 郑志兵; 肖军海; 任珅; 李松; 军事医学科学院毒物药物研究所; 军事医学科学院毒物药物研究所 北京;摘要:目的:建立高效液相色谱法测定新药盐酸纳美芬及其注射剂的含量及其有关物质。方法:采用迪马公司C18钻石色谱柱(250 mm×4.6 mm,5μm);流动相:乙腈-0.05 mol·L-1的磷酸缓冲液(20:80),其中1 000 mL缓冲液中含有7.8 g磷酸二氢钠和2 mL的三乙胺,用85%的磷酸调节pH为4.2±0.02;流速:1.0 mL·min-1;检测波长为210 nm。结果:HPLC法测定的线性范围为21-126μg·mL-1,r=1.000,最低检测限为0.2 ng,本方法的重复性和精密度良好(RSD2%),平均回收率为99.30%-99.42%。结论:采用HPLC法测定盐酸纳美芬及其注射液的含量和有关物质,方法简便,结果准确。http://ng1.17img.cn/bbsfiles/images/2012/07/201207161705_377929_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207161705_377930_2379123_3.jpg
我测定元宝枫和雪松的挥发物成分及含量选用3-壬酮,浓度为原液(99%,密度0.82),稀释100倍后,不合适,在稀释液基础上又稀释2倍,这样浓度应该是4.1mg/ml吧。元宝枫进样2ul,出峰较好,相对含量在5%左右;雪松在5ul,出峰较好,相对含量7%。根据内标物计算成分公式:含量mg/g=(组分峰面积/内标物峰面积)*(内标质量mg/样品量g)我理解的内标物质量mg,是进样量*浓度样品量g,是我选择的样品的质量数(叶片1og).如果这样理解的话,内标物的进样量不是很有关系吗?但是我如果用4.1mg/ml浓度在元宝枫中进样5ul的话,内标物占的比例很大35%左右,如果这样计算的话,与2ul的差别是数量级上的。我的上述理解对吗?现在很迷惑。查的资料用的浓度基本在0.41mg/ml的数量上。所以我很迷惑,如果我用4.1的进样2ul合理吗?看了很大资料,发现非化学的专业的,好像很多内标物浓度及计算都不怎么靠谱。如果我认为内标物不准确的话,我直接用峰面积和相对含量对各组分进行比较,应该也是可行的吧?谢谢!
应符合中国药典2000年版附录V D高效液相色谱法及附录XIX A药品质量标准分析方法验证的有关规定。若另有规定者应于正文中写清楚,如拖尾因子,附录中规定应在0.95~1.05,某品种若不能达到,可另制订合适指标。 一. “含量测定”可按下述格式表述 多数抗生素类药品的含量测定采用的是HPLC-紫外检测器法,其书写格式包括以下内容: (一)照高效液相色谱法(附录V D)测定。 (二)色谱条件与系统适用性试验:包括以下内容:色谱柱填料种类,流动相组成及配制方法,流速,检测波长,柱理论板数,拖尾因子,分离度测定溶液配制方法及分离度要求,进样量,数次进样对照品溶液应达到的精密度等。 注:1. 如果理论板数对分离度及定量测定结果影响不显著时可省略。 2 如果色谱峰的对称性较好,可不规定拖尾因子,但对称性差的品种应规定拖尾因子。 3. 考核分离度的方法有: (1)用已知杂质对照品,测定其与主组分峰的分离度,如头孢克洛含量测定时规定头孢克洛与d-3异构体对照品混合溶液的分离度应符合规定。 (2)杂质对照不能获得的情况下,可通过样品的适当处理使降解,测定降解物与主组分峰的分离度,如头孢呋辛钠含量测定中,取对照品溶液在60℃水浴中加热10分钟,冷却,使部分头孢呋辛转变为去氨基甲酰头孢呋辛后,测定头孢呋辛峰与去氨基甲酰头孢呋辛峰的分离度。 (三)对照品溶液的制备:应写清楚对照品溶液制备所用溶剂及溶液浓度。若储备液与测定液溶剂(稀释液)有差异时,应分别写清楚配制方法。 (四)测定法:应写清供试品溶液的配制方法。对于制剂若由于辅料会影响取样均一性者,还应规定合适的取样量。对于溶液稳定性差的品种,应写清“临用前配制”或“供试液应冷处保存”等。含量测定溶液浓度的确定,除了应在线性范围内外,应以规定的进样量测定的峰面积积分值在6位数以上为宜。 另外,也有一些无特征紫外吸收物质,例氨基糖苷类抗生素,可以采用HPLC-蒸发光散射检测器(Evaporative Light-Scattering Detector, ELSD)法测定含量。当采用该法测定含量时,书写格式可参考上述HPLC-紫外检测器法,但应注意以下几点: (1)“色谱条件与系统适用性试验”项:液相色谱条件中应注明流速,因为流速大小直接影响ELSD的检测器条件;检测器条件中应注明漂移管温度,载气流速。系统适用性试验一般要包括数次进样对照品溶液应达到的精密度(RSD),以确保检测器的稳定性(一般来说,该法要求RSD不得过2%)。 (2)“对照品溶液的制备”项:由于ELSD的响应值(A)与进样浓度(C)间呈指数关系(A=aCb,a,b均为常数),在采用该法测定含量时,计算方法宜采用随行校正曲线法(lgA=algC+b,a,b分别为校正曲线的斜率、截距),故对照品溶液的浓度至少应包括高、中、低3个不同浓度,浓度设计以能覆盖供试品溶液浓度为宜。 (3)“供试品溶液的制备与测定”项:同HPLC-紫外检测器法,但计算方法采用的是随行校正曲线法,即供试品浓度是根据供试品溶液中主峰峰面积和随行校正曲线(lgA=algC+b)来确定的,具体计算方法可参见“3 项撰写细则”一章中的“1.7 组分检查或纯度检查”部分。 二. 有关物质测定* (一) 有关物质测定的色谱条件与含量测定相同者,可参照中国药典2000版中头孢呋辛钠有关物质检查法表述。但其中有如下几点不完美: 1. 供试品溶液浓度低于含量测定时供试品浓度,有可能影响杂质的检测,应适 当提高。 2. 结果评价应以加校正因子或不加校正因子的主组分自身对照法计算出单个最大杂质或表观含量大于0.1%含量以上的杂质总和的含量或表观含量,而不以单个最大杂质或杂质峰面积总和不得大于对照品溶液主峰峰面积的多少倍表示。 (二) 有关物质测定的色谱条件与含量测定不同者,可参照中国药典2000年版中头孢克洛有关物质检查法表述,但其中有如下表述方法需作修改。 1. 流动相梯度洗脱方式以列表方式表述如下: 流动相A与流动相B按下表变换方式进行洗脱: 时间(分) 流动相A(%,V/V) 流动相B(%,V/V) 0~30 95%®75% 5%®25% 30~45 75%®0% 25%®100% 45~55 0 100% 55~56 0®95% 100%®5% 56~71 95% 5% 2. 结果评价方式同上,应计算出单个最大杂质含量或表观含量大于0.1%以上的杂质总和的含量或表观含量。 (三)有关物质测定中明确规定对照品溶液数次进样后相对标准偏差的限度,并以数次进样的平均值计算出各有关物质的量。 (*注:最低检出限或最低定量限的确定应按照中国药典的要求进行验证,以保证结果的可*性。)

【作者中文名】汪涛; 邓树海; 徐丽洒; 万发里;【作者英文名】WANG Tao1; DENG Shu-hai1*; XU Li-sa2; WAN Fa-li1(1.School of Pharmacy; Shandong University; Jinan 250012; China; 2.School of Pharmacy; Qingdao University; Qingdao 266021; China);【作者单位】山东大学药学院; 青岛大学药学系; 山东大学药学院 济南; 山东青岛;【摘要】目的建立HPLC测定扎托洛芬片的含量及有关物质的方法。方法色谱柱为Diamonsil C18柱(4.6 mm×150 mm,5μm),流动相为乙腈-水-醋酐(60∶40∶0.5),检测波长332 nm。结果扎托洛芬在30~70 mg.L-1内线性关系良好(r=0.999 9),平均回收率为99.7%(RSD=0.74%)。结论本方法操作简单,分离效果好,灵敏度高,结果准确可靠。http://ng1.17img.cn/bbsfiles/images/2012/08/201208271749_386604_2379123_3.jpg
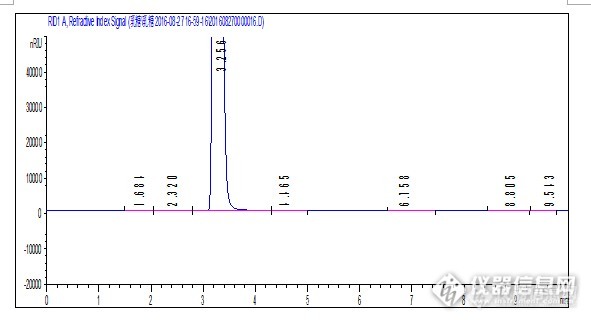
HPLC-RID法测定乳糖含量和有关物质 前言: 乳糖是我们制药领域,最常用的辅料之一,我们药用乳糖对其含量和杂质的限量有极其严格的要求。以下就是我们使用《中国药典》2015年版四部标准方法,对购入的一批药用乳糖的含量和有关物质进行分析的过程 ,与大家一起分享,希望各位老师多多指导。一、【含量测定】仪器型号电子分析天平:赛多利斯 BT125D型十万分之一电子分析天平 液相色谱仪:Agilent 1260 四元梯度液相色谱仪色谱条件:色谱柱:氨基柱(上海月旭 拓扑 250*4.6) 检测器:示差折光检测器 检测器温度:40℃ 流速:1.0ml/min 柱温: 45 ℃ 流动相: 乙腈-水(70 :30 ) 进样量: 10μl 对照品乳糖、蔗糖对照品使用来源: 中国食品药品检定研究院 系统适用性试验: 精密称取乳糖对照品10.15mg、蔗糖对照品10.21mg,置10ml容量瓶内,用水稀释至刻度摇匀。乳糖峰与蔗糖峰的分离度应大于1.5,理论塔板数以乳糖峰计不得低于5000。对照品溶液的制备: 精密称取乳糖对照品两份分别为10.10、10.08mg置10ml容量瓶内,用水稀释至刻度摇匀,即得。供试品溶液的制备:取本品适量,精密称定,两份分别为10.05、10.03mg, 置10ml量量瓶内,用水稀释至刻度摇匀,即得。检测结果:http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669315_2204446_3.png 系统适应性试验: 乳糖、蔗糖分离度均大于1.5,乳糖理论塔板数大于5000http://ng1.17img.cn/bbsfiles/images/2017/10/2016093023083649_01_2204446_3.png 乳糖对照品色谱图http://ng1.17img.cn/bbsfiles/images/2017/10/2016093022320255_01_2204446_3.png 乳糖供试品色谱图通过外标法计算本批按无水物计算含量为98.9%,在药典规定的98.0%至102.0之间,符合标准规定。二、【有关物质】色谱条件同含量测定项下方法测定供试品溶液的配制:取本品,精密称定 1.0023g,置10ml量瓶中,用适量水使溶解,并稀释至刻度,摇匀,过滤,即得。对照品溶液的配制:精密量取供试品溶液1ml,置100ml量瓶中,用水稀释至刻度,摇匀,过滤,即得。有关物质供试品色谱图如下:http://ng1.17img.cn/bbsfiles/images/2017/10/2016093022502870_01_2204446_3.png通过计算个杂质峰面积之和小于对照峰面积的0.5倍,小于0.5%。 总结:HPLC-RID测定乳糖含量和有关物质,样品处理简单,要注意的是RID平衡很关键,保证检测器外部环境的稳定很受到重要。RID为通用型检测器,容易受到气体流动和温湿度的影响。另外色谱柱的选择也很重要,一般的柱子乳糖理论踏板数上5000很容易,上海月旭拓扑氨基柱优越的性能为检测节省很多宝贵的工作时间。
有谁做过阿莫西林含量和有关物质,药典中提到色谱图应与标准图谱一致,标准图谱指的是什么,哪里有,含量中提到用系统适用性对照品,哪里可以买到?

【作者】 郝万红; 孙红梅;【Author】 Hao Wanhong1,Sun Hongmei2 (1.Pharmaceutical Factory,Shandong Wanjie High Tech.Stock Co.,Ltd.,Zibo,Shand ong,China 255213;2.Wanjie Hospital of Zibo,Shandong Province,Zibo,Shandong,China 255213)【机构】 山东万杰高科技股份有限公司制药厂; 山东淄博万杰医院 山东淄博255213; 山东淄博255213;【摘要】 目的用高效液相色谱法(HPLC法)测定去氧氟尿苷的含量及其有关物质。方法采用DiamonsilC18柱(250mm×4.6mm,5μm),流动相为乙腈-水(1∶1),检测波长为269nm,流速为0.8mL/min。结果去氧氟尿苷质量浓度在10.0~200.4μg/mL范围内与峰面积线性关系良好,r=0.9999(n=5),平均回收率为100.2%,RSD=0.14%。结论HPLC法简便、准确,专属性好,灵敏度高,可用于去氧氟尿苷含量及有关物质的测定。 更多还原【Abstract】 Objective To establish HPLC method for determination of doxiflurid in e content and its related substances.Methods The column was Diamonsil-C18 colum n (250 mm×4.6 mm,5 μm).The mobile phase was acetonitrile-water (1 ∶1).The detection wavelength was 269 nm.The flow rate was 0.8 mL/min.Results There was a good linear relationship with the range of 10.0-200.0 μg/mL for determinati on of the doxifluridine’s content,r=0.999 9(n=5).The average recovery rate was 100.2%,RSD=0.14%.Conclusion The method i... 更多还原【关键词】 去氧氟尿苷; 含量测定; 有关物质; 高效液相色谱法; 【Key words】 doxifluridine; content determination; related substances; HPLC; http://ng1.17img.cn/bbsfiles/images/2012/08/201208271629_386499_2352694_3.jpg

【作者】 满凤; 安穗伟; 梁北梅; 彭锋; 刘学斌; 李发美;【Author】 Man Feng1,An Sui-wei2,Liang Bei-mei2,Peng Feng2,Liu Xue-bin2 and Li Fa-mei1 (1 School of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016; 2 Guangzhou Pharmaceutical Industrial Research Institute,Guangzhou 510240)【机构】 沈阳药科大学药学院; 广州市医药工业研究所;【摘要】 目的建立测定左旋尤利沙星含量及有关物质的高效液相色谱方法。方法采用DiamonsilC18柱(250mm×4.6mm,5μm),以乙腈-0.2%三乙胺和0.1%庚烷磺酸钠水溶液(用磷酸调节pH2.5)(25:75)为流动相,流速为1.0ml/min,检测波长为275nm,柱温为35℃。结果主峰能与相邻杂质峰达基线分离,左旋尤利沙星的浓度在10~200μg/ml范围内线性关系良好,相关系数为0.9996,最低检测限为0.2ng。结论该法简便、准确,灵敏度高,专属性强,可以用于左旋尤利沙星的含量及有关物质的测定。http://ng1.17img.cn/bbsfiles/images/2012/07/201207181205_378449_1761902_3.jpg
目的:建立盐酸舍曲林片含量及有关物质测定的高效液相色谱方法。方法:色谱柱为Diamonsil C18(4.6mm×200mm,5μm);流动相为甲醇-0.2%三乙胺溶液(用磷酸调到pH4.0)(60∶40),流速1.5mL/min;检测波长225nm。结果:进样量在2.49~19.94μg范围内,与峰面积线性关系良好,平均回收率为99.55%,RSD为0.60%(n=9)。结论:该方法简单、快速、准确,重现性及专属性好,可用于盐酸舍曲林片的质量控制。
365个日日夜夜,1年接着1年,1095个日日夜夜。终于结束了。。。我们的项目品种一般考察至36个月,该品种终于考察完成了,相应色谱柱也要转移到其他的品种上了,继续发挥他们的作用。。。其中的36个月中,在考察中,其杂质谱的变化以及含量变化最牵动人心,其中有样品本身的变化以及设备仪器性能的不稳定,增加了很多的工作量。最终,利用月旭公司的色谱柱完成了检测任务。1095,终点即起点篇(一)http://ng1.17img.cn/bbsfiles/images/2013/10/201310240902_472574_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472575_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472576_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472577_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472578_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472579_1621890_3.gif0月全检色谱图:http://ng1.17img.cn/bbsfiles/images/2013/10/201310251438_472923_1621890_3.gif0月和36月色谱图对比情况:http://ng1.17img.cn/bbsfiles/images/2013/10/201310251439_472924_1621890_3.gif备注:色谱图从上至下名称为0月全检含量测定供试液图、长期稳定性考察36月含量测定供试液图。http://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472581_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240903_472582_1621890_3.gif0月全检色谱图:http://ng1.17img.cn/bbsfiles/images/2013/10/201310251431_472918_1621890_3.gif0月和36月色谱图对比情况:http://ng1.17img.cn/bbsfiles/images/2013/10/201310251432_472919_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310240904_472583_1621890_3.gif结论:以前的色谱柱由于拖尾严重、分离度未达到1.5(主峰前的杂质),不适合检验本品的有关物质,但改用月旭公司的色谱柱能满足要求且杂质的个数和相对保留时间一致(杂质谱一致),证明了月旭公司的色谱柱耐用性好(通用性强),和其他公司比较无明显差异,这样有利于品种有关物质色谱柱的选择。其研究内容详见附件。

高效液相色谱法测定盐酸甲氯芬酯分散片含量及有关物质周岐勋 李健和 徐幸民 彭六保 曹俊华 罗霞(1.湖南省湘西自治州人民医院药剂科,湖南吉首416000;2.中南大学湘雅二医院药剂科,湖南长沙410011)【摘要】目的:建立盐酸甲氯芬酯分散片含量与有关物质测定的高效液相色谱分析方法。方法:采用Diamonsil C18柱(250 mmx4.6 mm,5um)。以乙腈_o.02 mol/ml磷酸二氢钾溶液(磷酸调pH值至4.0)(33:67)为流动相,检测波长为225 nm,流速为1.0 ml/min,柱温为25℃,进样量为20ul。结果:有关物质测定中降解产物与样品主峰能有效分离,分离度符合要求。盐酸甲氯芬酯最低检出量为2.0 ng。3批样品中有关物质的平均含量为1.56%。样品浓度在5.2—20.8斗∥ml的范围内与峰面积呈良好的线性关系(r=1.000),平均回收率为99.26%(RSD=I.12%)。结论:本检测方法专属性强.结果可靠。重复性良好,可用于盐酸甲氯芬酯分散片的质量控制。【关键词1盐酸甲氯芬酯分散片;有关物质;含量测定;高效液相色谱法http://ng1.17img.cn/bbsfiles/images/2012/07/201207232327_379311_2355529_3.jpg
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=72386]反相高效液相色谱法测定依诺沙星原料含量与有关物质[/url]
还有测一个物质的含量一定要标样吗

【作者】 税庆华; 韩保萍; 王玉玲; 张世磊; 于盛茂;【Author】 SHUI Qing-hua1,HAN Bao-ping1,WANG Yu-ling1,ZHANG Shi-lei1,YU Sheng-mao2(1. Shandong Bausch﹠Lomb Freda Pharmaceuticals Co.,Lid.,Jinan 250014,China;2. Institute of Biopharmaceuticals of Shandong Province,Jinan 250014,China)【机构】 山东博士伦福瑞达制药有限公司; 山东省生物药物研究院;【摘要】 目的建立HPLC测定盐酸四氢唑啉滴眼液含量的方法。方法采用Diamonsil C18(4.6mm×150mm,5μm)色谱柱;以甲醇-醋酸盐缓冲液(40:60)为流动相,检测波长为235nm。结果盐酸四氢唑啉质量浓度在9.94~497.0mg.L-1内与峰面积呈良好线性关系(r=0.9999);最低检测限为0.22ng;平均回收率为99.86%,RSD为0.23%。结论本方法灵敏度高,方法操作简便,可满足本品含量及有关物质的测定。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208271710_386575_2379123_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP