做有关物质分析时,流动相里需要用到盐,这个盐要用优级纯的吗?
有做过申报的朋友吗?化学药的有关物质分析方法中,如果空白在主峰处有一个很小很小的峰,只比基线稍微高出一点,这样的方法可以吗?同样,如果这个小峰出在和主要杂质峰相同的地方呢?
做有关物质分析时,空白溶剂在13min处有一峰面积约为7的杂峰,分析样品时,13min处的杂峰面积约20,这说明空白有干扰吗?方法是按药典的方法,应该不会有什么问题吧
能不能请做过丙二醇有关物质的老师帮忙解决一下丙二醇有关物质的图谱分析和计算问题?
摘要:本文介绍了在对有关物质检查所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。 关键词:有关物质检查 分析方法验证 可接收标准 药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。
RT:有关物质需不需要分析方法验证有关物质是按自主成分对照法来分析的
[align=right][b]SGLC-LC-338[/b][/align][b]摘要:[/b]本文建立了头孢呋辛钠有关物质分析的HPLC方法。参照2020版《中国药典》中色谱条件,采用色谱柱ShimNex HE C8分析头孢呋辛钠有关物质,结果显示,去氨甲酰头孢呋辛与头孢呋辛分离度大于3.0,且主峰与后相邻杂质峰基线分离,满足《中国药典》要求。此方法可为头孢呋辛钠有关物质分析提供参考。。[b]关键词:[/b]头孢呋辛钠 有关物质 ShimNex HE C8 HPLC[b]1. 实验部分1.1 实验仪器及耗材[/b]Shimadzu LC-40D高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url];色谱柱:ShimNex HE C8 (5 μm,4.6×250 mm;P/N:380-01241-09);纯水机:PR-FP-0120α-MT1(+ 60L水箱 + 取水器)SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34001-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 系统适用性溶液的制备[/b]取头孢呋辛对照品适量,加水溶解并稀释制成每1 mL含0.5 mg的溶液,置60℃水浴放置30分钟,放冷,使头孢呋辛部分转化为去氨甲酰头孢呋辛。[b]1.3 分析条件[/b]色谱柱:ShimNex HE C8 (5 μm,4.6×250 mm;P/N:380-01241-09)柱温:30℃检测波长:273 nm流速:1.0 mL/min进样量:20 μL流动相:A: 醋酸盐缓冲液(取醋酸钠0.68 g,冰醋酸5.8 g,加水稀释成 1000 mL,用冰醋酸调节pH值至3.4) B:乙腈梯度程序如下:[img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-01.png[/img][b]2. 实验结果[/b]按照上述色谱条件(1.3)进行采集,系统适用性溶液色谱图如下:[b]系统适用性溶液[/b][img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-02.png[/img][b]系统适用性放大图[/b][img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-03.png[/img][img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-04.png[/img][img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-05.png[/img][b]重现性[/b]系统适用性溶液重现性[img]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-LC-338-06.png[/img][b]3. 结论[/b] 本文建立了头孢呋辛钠有关物质分析的HPLC方法。参照2020版《中国药典》中色谱条件,采用色谱柱ShimNex HE C8分析头孢呋辛钠有关物质,结果显示,去氨甲酰头孢呋辛与头孢呋辛分离度大于3.0,且主峰与后相邻杂质峰基线分离,满足《中国药典》要求。此方法可为头孢呋辛钠有关物质分析提供参考。
有关物质分析时,关于积分现在有两种说法:1.供试液杂质峰面积在主峰面积的0.05%以上才积分。2.大于检测线的都积分。应该哪个更合适些?或有没有其他说法?
最近在做一个产品的有关物质分析,用的是HPLC法,质量标准为欧洲药典标标。具体的分析条件如下:色谱柱为XBP-C1柱。色谱柱长0.25m,内径4.6m;三甲基硅烷键合硅胶做为填充剂,流动相为水和甲醇体系;检测波长254nm;流速为2.5ml/min;现在的问题在几个小时内重复进样,主峰的保留时间波动较大,最小为6.078min,最大为7.758min,请问怎么会出现这种情况,是不是与柱子的流失有关系,注:我们用的是新柱子。
最近在做一个产品的有关物质分析,用的是HPLC法,质量标准为欧洲药典标准。具体的分析条件如下:色谱柱长0.25m,内径4.6m;三甲基硅烷键合硅胶做为填充剂,流动相为水和甲醇体系;检测波长254nm;流速为2.5ml/min;现在的问题是流速太大,导致柱压太高,泵受不了,如何解决这个问题?与柱子的关系大吗?
请问诸位专家: 高效液相分析图谱的有关物质怎样积分?是以手动积分还是自动积分为准??自动积分时,积分很不合理或者很不规范怎么办??
分析柱的柱效(理论塔板数)是不是和被分离的物质有关?是不是同一根柱子分离不同化合物的塔板数不一样?那一般说柱子的柱效是用什么测出来的??谢谢,可能比较傻的问题。
阿奇霉素有关物质分析方法对色谱填料的要求目前2005版中国药典对阿奇霉素相关物质的测定采用薄层色谱法(TLC),对含量的测定是采用微生物检定法,方法的专属性不好。USP以及欧洲药典现行分析方法是采用液相色谱法。
流动相为0.2%甲酸-乙腈 20-80,流速1ml/min,CAD检测器分析一个紫外吸收200nm左右的化合物。因为要进质谱对有关物质进行定性,又避免甲酸,乙酸,乙酸铵等在200nm处的影响,所以选择用CAD检测器。以上述条件分理处三个杂质,但是主成分峰形不好,尝试在0.2%甲酸水中加入了5mM甲酸铵,10mM乙酸铵等,发现峰型改善了,但是杂质都没了,这是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008202147293471_645_4037032_3.png[/img]
药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至一定的浓度范围内配制6份浓度不同的供试液,分别测定该杂质峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。 3.精密度 1)重复性 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于15%。 2)中间精密度 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于20%。 4.专属性 可接受的标准为:空白对照应无干扰,该杂质峰与其它峰应能完全分离,分离度不得小于2.0。 5.检测限 杂质峰与噪音峰信号的强度比应不得小于3。 6.定量限 杂质峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±5nm、流速相对值变化±20%以及采用三根不同批号的色谱柱进行测定时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:各杂质峰的拖尾因子不得大于2.0,杂质峰与其他成分峰必须达到基线分离;各条件下的杂质含量数据(n=6)的相对标准差应不大于2.0%,杂质含量的绝对值在±0.1%以内。 8、系统适应性 配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%,保留时间的相对标准差应不大于1.0%。另外,杂质峰的拖尾因子不得大于2.0,理论塔板数应符合质量标准的规定。 9.溶液稳定性 按照分析方法分别配置对照品溶液与供试品溶液,平行测定两次主成分与杂质的含量,然后将上述溶液分别贮存在室温与冰箱冷藏室(4℃)中,在1、2、3、5和7天时分别平行测定两次主成分与杂质的含量。 可接受的标准为:主成分的含量变化的绝对值应不大于2.0%,杂质含量的绝对值在±0.1%以内,并不得出现新的大于报告限度的杂质。
此问题代mjxyz版友发帖:请教一下:现在在做一个制剂的有关物质分析方法验证,其专属性,做了破坏试验,现在发现破坏试验的条件很不好摸索,没有规律;酸碱的浓度时间变化对主成分的峰面积下降 不成任何规律。所以想请教是否我自己理解错误,1.破坏降解率是以API的峰面积下降的比率为标准,还是以API计算后的含量下降为准?2.酸 碱破坏完需要加入对应的碱和酸中止破坏吗?3.因供试品中存在一个已知杂质和若干未知杂质,那专属性试验-中未知杂质要怎么考虑?是只设定个鉴别阈值?对未知有没有分离度的要求?4.破坏试验的样品配制是以含量测定方法的步骤配?还是以有关物质检查的稀释流程?
药典方法分析乙醇的有关物质的毛细管柱,用什么型号的最好?
有关物质分为已知杂质和未知杂质,如果是未知杂货需要做的有:1.系统适用性试验2.专属性3.检测限4.溶液的稳定性5.耐用性已知杂质需要做的有:1.系统适用性2.准确度3.精密度4.专属性5.定量限6.线性7.溶液的稳定性8.耐用性问题1:其中已知杂质的系统适用性是配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%。那么未知杂质的系统适用性试验应该如何进行呢?问题2:未知杂质中检测限的测定,因无法获得杂质来源,是否应该用供试品溶液测定,观察杂质峰与噪音响应的比例?问题3:如果有多个已知杂质,且都是用同种HPLC条件分离测定的,那上述7个验证项目(除专属性外)是否都需要用不同的杂质配制溶液后重复检测呢?
目前我们在开发对羟基苯甘氨酸羟邓盐有关物质分析方法遇到了些困难,采用Waters,Xbidge BEH Shiled RP18,250*4.6mm,5um的色谱柱,流动相为乙腈与磷酸盐缓冲液混合,且pH调节至6.80。主峰几乎没有保留,峰尾部不能落回原点基线水平,基线后部分漂移较大。该物质好像容易水解。各位老师能否给些建议。
要写有关有机脂肪酸类物质的色谱分析方法研究进展的论文,找了好久都没有,请各位指点一下有哪些相关的书和文章,急!拜谢!
在开发GC分析方法时柱温的选择是否至少应该高于分析物质的沸点?柱子的选择是与物质的极性还是与物质的沸点或分子量有关?分析杂环化合物一般选什么样的柱子?
请问怎样着手进行一个药物的有关物质研究?我研究的药物虽属有机物,但无紫外吸收。进行其有关物质研究需考虑哪些因素?需查阅哪些方面的资料?谢谢有经验的人指点帮忙。谢谢!
各位老师好!目前我们这边在开发 对羟基苯甘氨酸邓钾盐有关物质的分析方法 ,但是尝试了很多条件都不行。主要问题是i主峰出峰后峰尾部分不能落回原点的基线水平,而且基线在后半部分漂移很大,干扰到杂质峰。目前用的流动相是乙腈-0.01mol/L磷酸二氢钾溶液,用氢氧化钾溶液调节pH至6.8;色谱柱是 Waters, Xbridge BEH [color=#3e3e3e]Shield RP18。样品信息和图谱在附件。哪位老师有好的意见还望不吝赐教,谢谢!(该物质似乎很容易水解)[/color]
做有关物质时,所以的杂峰都要积分吗?还是特别小的不用积分呢?
用DAD做有关物质分析,很多杂质都看不到了,怎么办呢?
有哪位高手做过 盐酸金刚烷胺 这种产品,它的有关物质这项指标用哪种色谱柱分析好些? 我用的是DB-54的 柱子
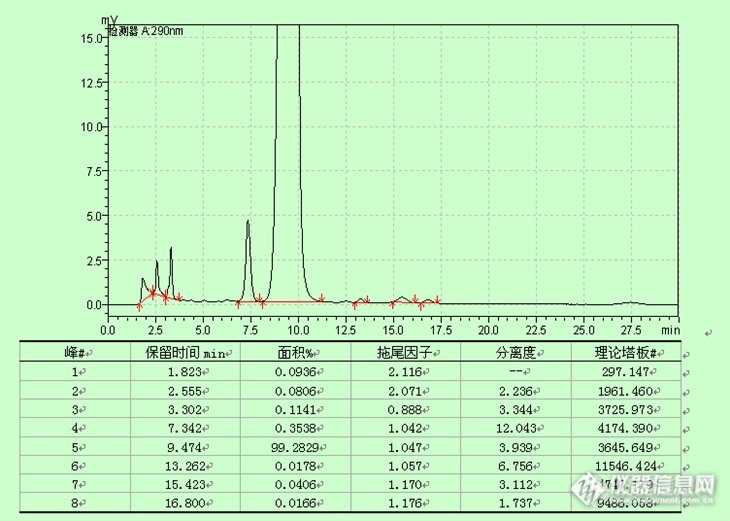
此篇文章,相对于杂质谱分析是个综述性质的。项目:有关物质试验条件及操作检查方法:HPLC法试验条件:色谱柱(柱长:250mm,内径:4.6mm,填料:C18,填料粒径:5μm)月旭色谱柱:SN:W10212097;PN:weL518425。UV检测器(检测波长:290nm)柱温:30℃流动相:0.05mol/L磷酸盐溶液(用0.05mol/L磷酸二氢钾溶液调节0.05mol/L磷酸氢二钠溶液pH值至7.0)-甲醇(40:60)流速:1.0ml/min运行时间:约30min具体试验操作:取含量测定项下的细粉适量(约相当于雷贝拉唑钠50mg),精密称定,置50ml量瓶中,加0.05mol/L氢氧化钠溶液20ml,超声溶解,放冷至室温,用甲醇稀释至刻度,摇匀,在3000rpm下离心10分钟,取上清液作为供试品溶液。精密量取供试品溶液1ml,用0.05mol/L氢氧化钠溶液-甲醇(2:3)稀释至100ml,作为对照溶液。精密量取对照溶液10μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的20%~25%;再精密量取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图至主成分色谱峰保留时间的2倍。对照溶液中的主峰面积As、供试品溶液中各杂质的峰面积Ai均通过自动积分测定,以各杂质峰面积与对照溶液主峰面积的比值计算得出各杂质的含量,总杂为各杂质和。计算公式:各杂质的量(%)=Ai/As杂质总量(%)=∑i1.专属性试验,主要是分析色谱条件能否满足分离出更多的杂质,以及色谱峰参数符合药典要求。有已知杂质更好,没有,就只能进行破坏产生杂质,分析汇总结果,列出杂质谱。一般做法就是以相对保留时间列表统计,然后再进行物理平衡,这样能从侧面验证,杂质检出的最大限量。举例:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292237_448395_1621890_3.png物料平衡,主要以响应值来进行平衡,如:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292239_448396_1621890_3.png杂质谱做出来了就要和原研上市品比较,主要考察杂质的个数以及对应情况,如:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292241_448397_1621890_3.png最好,直观比较,用工作站把各色谱峰进行比较,如:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292242_448398_1621890_3.png其他项目,我在这个月的原创里面谈了很多,如检出限定量限、精密度、稳定性等,就不谈了。2.稳定性考察的杂质谱比较,主要考察新增杂质个数及含量变化,若样品不稳定,也同条件下进行上市品考察比较,如:【检查】有关物质 本品有关物质检查采用高效液相色谱法,并对方法进行了方法学验证,验证试验结果均符合要求。本品流动相选择试验结果显示,以0.05mol/L磷酸盐溶液(用0.05mol/L磷酸二氢钾溶液调节0.05mol/L磷酸氢二钠溶液pH值至7.0)-甲醇 (40:60)为流动相能满足本品有关物质检查要求;根据本品专属性试验统计结果,将检测波长选择为290nm。限度确定:经过加速试验和长期试验,本品在加速条件为温度为40±2℃、相对湿度为75±5%加速试验条件下,考察至2个月时,本品有关物质变化情况为单杂在0.7%~1.7%,总杂在0.9%~5.4%(总杂限度为3.5%);温度为30±2℃、相对湿度为65±5%加速试验条件下,考察至6个月时,本品有关物质变化情况为单杂在0.7%~1.3%,总杂在0.9%~1.9%;长期试验条件下考察至18个月,本品有关物质变化情况为单杂在0.7%~0.9%,总杂在0.9%~1.3%。http://ng1.17img.cn/bbsfiles/images/2013/06/201306292246_448399_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292247_448400_1621890_3.png再直观作图,杂质谱统计也要做就不累述了。作图:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292250_448401_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292250_448402_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292252_448403_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292253_448404_1621890_3.png这样作图就很直观了,审批的老师看起来也不吃力,就有好运哈。说了半天,整张美图看看:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292257_448405_1621890_3.png总结:1.明确研究项目内容及要点,结合ICH以及相关国内的指导原则,规划试验项目及进展;2.每个项目分解后总结,就如涓流成溪一样,说明您要表达的试验意图,最好表图结合直观表达;3.开展一个项目,就如有关物质,要准备好至少两根同型号的色谱柱,还有其他主流品牌的,特殊色谱柱除外;这样有几大好处,如杂质谱好归属
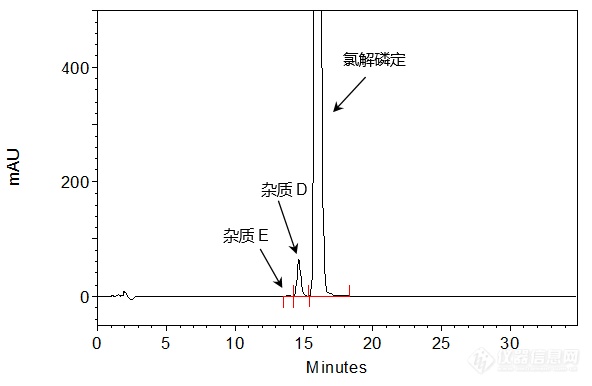
[align=center][b]注射用氯解磷定的有关物质分析[/b][/align]氯解磷定注射液是有机磷中毒解毒药,对急性有机磷杀虫剂抑制的胆碱酯酶活力有不同程度的复活作用,用于解救多种有机磷酸酯类杀虫剂的中毒。[align=center][img=,105,92]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656260751_676_2222981_3.gif!w105x92.jpg[/img][/align][align=center]氯解磷定[/align][align=center]2-Pyridinealdoxime methochloride[/align][align=center]C7H9ClN2OM.W.: 172.6[/align][align=center][/align]客户提供了注射用氯解磷定样品,希望本实验室依据客户指定色谱条件筛选合适的C[sub]18[/sub]色谱柱,以实现氯解磷定注射液样品的有关物质分析,满足氯解磷定主峰同相邻杂质峰以及各杂质峰间的基线分离要求。首先,尝试使用[b][color=red]中等极性的普适型色谱柱——[/color][color=red]CAPCELL PAK C[sub]18[/sub] MGII[/color][/b],依据客户所提供的色谱条件(流动相含十二烷基硫酸钠和二乙胺)对氯解磷定注射液样品进行分析。如图1,[color=#2E74B5]氯解磷定主峰保留时间为[/color][b][color=#2E74B5]16 min[/color][/b][color=#2E74B5],主峰与峰前杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.33[/color][/b][color=#2E74B5],杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]与杂质[/color][color=#2E74B5]E[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]1.64[/color][/b][color=#2E74B5],均能得到良好分离结果。通过对相对保留时间进行计算,所得结果满足客户[/color][color=#2E74B5]SOP[/color][color=#2E74B5]要求。[/color][color=#2E74B5][/color][align=center][img=,592,386]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656248670_1537_2222981_3.png!w592x386.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18[/sub] MGII分析所得色谱图[/align][align=center][/align][align=center][/align][align=center][/align][align=center][img=,587,389]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656253460_3177_2222981_3.png!w587x389.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18[/sub] MGII分析所得色谱图放大图[/align]注:峰上标数字由下至上依次为分离度、理论塔板数与保留时间。[img=,424,282]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656270060_717_2222981_3.png!w424x282.jpg[/img]就上述实验结果与客户沟通,客户反映希望使主峰的保留时间在20min左右。为满足客户需求,我们在上述实验条件基础上,将柱温由初始条件的40°C降至30°C进行分析,发现主峰保留时间为17 min左右;为进一步增强其保留,我们将色谱柱更换为[b][color=red]极性更高的[/color][color=red]CAPCELL PAK C[sub]18[/sub] AQ[/color][color=red]色谱柱[/color][/b],如图3,[color=#2E74B5]氯解磷定主峰保留明显增强,保留时间约[/color][b][color=#2E74B5]20 min[/color][/b][color=#2E74B5],主峰与峰前杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.88[/color][/b][color=#2E74B5],杂质[/color][color=#2E74B5]D[/color][color=#2E74B5]与杂质[/color][color=#2E74B5]E[/color][color=#2E74B5]之间分离度为[/color][b][color=#2E74B5]2.72[/color][/b][color=#2E74B5],能够得到良好的保留与分离结果。[/color][align=center][img=,599,388]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656255480_6169_2222981_3.png!w599x388.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18[/sub] AQ分析所得色谱图[/align][align=center][img=,597,361]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656258121_3421_2222981_3.png!w597x361.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18[/sub] AQ分析所得色谱图放大图[/align]注:峰上标数字由下至上依次为分离度、理论塔板数与保留时间。[img=,499,322]http://ng1.17img.cn/bbsfiles/images/2018/07/201807181656269602_541_2222981_3.png!w499x322.jpg[/img]综上实验结果,使用大曹色谱CAPCELL PAK系列色谱柱中的第一选择——中等极性的普适型色谱柱CAPCELL PAK C18 MGII和能在纯水条件下稳定使用的高极性色谱柱CAPCELL PAK C18 AQ进行分析,均能实现注射用氯解磷定的有关物质分析,并能满足氯解磷定主峰同其相邻杂质及各杂质峰间的基线分离要求,客户可根据实际需求进行选择。[align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
有关物质研究一直是药物药学研究和评价中的重点和难点。随着我们对有关物质认识的不断深入,对有关物质的技术要求也在不断提高,其中对有关物质进行定性(鉴定)研究已经成为有关物质研究技术要求的重要组成部分,受到广泛关注。但目前有关物质的定性研究仍然是国内有关物质研究中的薄弱环节,研究水平与国外先进国家相比有较大的差距,也限制了我国药品研究水平和药品质量提高,需要引起国内研发者的充分重视,主动进行深入研究,提高研究水平。 对药物中的有关物质进行定性研究具有重要意义,通过对有关物质的定性研究,我们可以获得有关物质的结构信息,分析其形成过程,合成时可设法避免该杂质的形成,或经纯化使之降至可接受的水平。另外,还可以通过检索毒性物质数据库获知该杂质的毒性数据,为其限度的确定提供有力的依据。同时,定性研究也是分析方法确定的重要参考,对贮藏条件的确定也有指导意义。如果不了解有关物质的结构,后续的研究将无法继续进行。 对药物中的有关物质进行定性分析也是我国药品研发与国际接轨的需要。按照ICH Q3A/Q3B指导原则以及2005年SFDA《化学药物杂质研究技术指导原则》的要求,对药物中超过鉴定限度的有关物质皆应明确其来源,并推测可能的结构。我国的药物研发要想与国际接轨,就必须在有关物质研究的完整性和规范性上符合相关的要求。 按照研究方法的不同,有关物质的定性研究可以分为理论推导法、直接测定法和间接测定法等,在实际的应用中,这几种方法常常结合使用,相互印证。一般而言,由于有关物质的定性研究不能像原料药的结构确证一样提供全面的信息,因而需要尽可能采用多种方法(直接测定法例外),提供尽可能多的信息和证据,否则,有可能得到错误的结论。下面对几种方法进行分别说明:
现在手上有一组氨酸原料药,几乎相当于纯品。老板要我做组氨酸的有关物质,除了其他的氨基酸,其他杂质是什么无从下手。DAD,CAD检测几乎没有杂质,进样量40ul才有点杂质峰出来。手上有一台q-e,不知道能否先直接分析组氨酸原料药里的组分,然后再寻找合适的液相条件开发有关物质的分析方法。由于原料药杂质含量太低了,但是的确有,实在不知道怎么进行了,哎







 我要推广仪器
我要推广仪器
 下载APP
下载APP