版友问题,做乙醇甲苯残留溶剂顶空进样不出峰,做其他的直接进样正常,会有什么原因呢?
二部药典中乙醇残留溶剂的检测关于乙醛这个单项,对照溶液(b)配置为加50ul乙醛、50ul无水甲醇用供试品溶液稀释至50ml,再吸取100ul至10ml.进样结果,将供试品溶液和对照溶液(b)的乙醛出峰面积带入计算,总是不合格!怀疑是乙醛的问题,不知道有没有遇到过这种情况?还有大家知不知道乙醛是怎么保存的,什么状态下能判定乙醛失效!还有乙醛充氮保护又是什么意思?
大家好,本人新手第一次测残留溶剂,外标法测水中无水乙醇。看了网上的方法,浓度范围100-400左右,但是拖尾因子一直在1.045附近,想调整拖尾小一点,但是又找不到好的方法。我的方法进样口150 检测器250 柱温70,11分钟,平衡温度70,40分钟,流量2,分流比10
各位大虾好,小第有一疑问盼大家给点意见。我做美国药典中的盐酸米托蒽醌中的残留溶剂时,按照厂家给的资料要检测其中的2-甲氧基乙醇,厂家给的方法为直接进样(水做溶剂)。但它的样品浓度为200mg/ml,按照盐酸米托蒽醌的溶解度看,样品是绝对不能完全溶解的,样品溶解后成米糊状,离心处理也处理不到上清液,进样发现样品出峰非常的难看,且不见残留溶剂峰。我们打算自己开发方法用顶空做,但美国药典467中建议2-甲氧基乙醇用直接进样法,各位大虾你有过内似的经历么,2-甲氧基乙醇用顶空法检测后,美国FDA能批准么?
[font=&]各位大神,我遇到顶空残留的问题,顶空进空气和水样都会有残留峰出现,而且测水峰面积比测空气大3-4倍,出峰时间在3.9和6.5分钟左右,但是进氮气就没有峰出现,顶空针,六通阀,切换阀,GC进样口,检测器口都洗过了,柱子也截了老化了,测试水和空气还是一样的峰[/font][font=&]空白溶剂是水 测乙醇残留 [/font][font=&]条件:恒温炉 80 样品流路 90 传输线105 [/font][font=&] 进样口 200 柱子程序升温50-220 [/font]检测器250 进样口压力42KPa 总流量8.1ml 柱流量 0.46ml 分流比10[font=&]GC是顶空和液体混用 液体进样测试的样品出峰面积有1亿多面积,也有1千多万的面积 [/font][font=&]最有疑问的是进了氮气就没有峰出现 [/font][font=&]现在怀疑分流流路和吹扫流路有残留[/font]且实验条件的流量偏小 但是测氮气也是一样的条件就没有峰就又矛盾了[font=&][/font][font=&] [/font][font=&] [/font]
本人菜鸟在做溶剂残留VOCs比对试验中,26种物质,混标溶液中有5种物质,分高浓度与低浓度两套标液,5种物质分别是甲醇、2-乙氧基乙醇、苯、环己酮、丁二酸二甲酯(按保留时间先后排列),其中甲醇、苯、丁二酸二甲酯均未出现异常,均为正偏离;但是2-乙氧基乙醇检测结果显示,高低两个浓度结果是所有实验室中的极小值,低浓度结果是离群,而环己酮的两个浓度结果为可疑,正偏离方向,求大神指教,应该如何着手分析,试验?
如题,残留溶剂做乙醇,丙酮,异丙醇,内标法,样品溶液丙酮和异丙醇都过了,就是乙醇每次过不了(精密度5%),已经做了3次,最好的一次乙醇也才5.99%。而标准品溶液精密度3次都在1.5%以下。标准品每次精密度都很低,是不是说明仪器方法其实问题不大?样品是药片,每次研磨成细粉,取700mg(药片每片350mg)放进顶空瓶,加4mL内标溶解,但其实压根就不溶,底下厚厚一层,我总觉得这样子样品里的乙醇挥发的都很不均匀,提出说是样品量减少,但领导不同意啊,请教各位大佬,给鄙人点建议吧,压力很大啊,不胜感激。
或者说 进行 甲醇 乙醇 三氯甲烷时 溶剂残留测定时 用什么检测条件方法比较好 ?
残留溶剂检查时,能用什么色谱柱或方法将乙醇和乙腈有效分离?谢谢!
我在做溶剂残留的时候,近出来的峰,乙醇峰老是包峰?请问怎么解决啊!
透明质酸钠是一种天然直链多糖,是由葡萄糖醛酸和乙酰氨基葡萄糖结合而成的双糖结构单元所组成,广泛分布于动物和人体皮肤、皮下组织、眼组织及关节滑膜组织、滑液等结缔组织,无种属差异。根据透明质酸钠结构单元的特性,对其进行深加工后制成的医用透明质酸钠凝胶是一种无种属特异性、无毒、溶解性能好、生物相容性良好的新型生物材料,被广泛用于多种眼科手术、防治外伤性或退变性骨关节炎、普通外科、妇产科等腹、盆腔手术、预防术后肠粘连和盆腔粘连及肌健、关节和神经手术预防组织粘连。透明质酸钠作为一种植人体内的医用生物材料,需要有与人体正常注射部位透明质酸钠更接近的分子量,以确保产品使用的安全性和有效性。光散射法是测定高聚物绝对分子量的方法,高分子溶液可视为不均匀介质,当光通过它时,入射光就会发生散射,且其散射光强度远高于纯溶剂,并且与高聚物的分子链形态、溶液浓度、散射光角度和折光指数增量(dn/dc)密切相关。因此由光散射法测得不同浓度的高聚物溶液在不同散射角下的散射光强数据后,即可求得其重均分子量。采用激光散射-凝胶渗透色谱联用法(LLS-GPC)得到分子量分布系数。色谱柱可采用SD805/806、TSK5000pw TSK6000,流动相推荐0.1 mol/L硝酸钠-0.02%叠氮化钠溶液。样品用上述流动相溶解并稀释至适宜浓度,用0.22pm滤膜过滤。折光指数增量(dn/dc)的测定:采用流动相稀释透明质酸钠凝胶至不同浓度梯度,在室温下,用激光检测同一波长测定。将色谱柱与激光散射仪,示差检测器连接,流动相冲洗至基线平稳后,取适量样品溶液进样,在规定流速、色谱柱温度条件下检测样品的分子量及分子量分布。检测完毕后,通过仪器配套的色谱分析软件确定样品的峰面积,输入dn/dc 值,根据软件要求设置其他相关参数,计算分子量及分子量分布系数并输出报告。透明质酸钠在提纯和干燥生产工艺中需要使用乙醇,而透明质酸钠是一种较难干燥的高分子材料,乙醇很难完全挥发掉,微量乙醇存在于透明质酸钠中将使产品在使用过程中对患者产生不同程度的刺激作用。因此,为了尽可能降低产品对患者的不良作用,同时也考虑到当前的生产工艺水平,对乙醇残留量进行合理限定是很必要的。本文介绍透明质酸钠中的乙醇残留量的检测方法,采用顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法使要测定的乙醇与其他组分分开,用氢火焰离子化检测器检测,并将得到的乙醇色谱峰与外标物得到的色谱峰相比较,行标规定医用透明质酸钠凝胶乙醇残留量应不大于400 ug/g。1.设备:配有氢火焰离子化检测器与毛细管柱系统的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],要求仪器对所测定的乙醇,在最低检测浓度下产生的信号大于仪器噪音的2倍。石英毛细柱:SE-30,50 m*0.53 mm*3.0 um或相同分离效果的其他色谱柱;柱温:500℃,保持2 min,以100℃/min的速率升至 160℃,保持 5 min;汽化、检测室温度:180℃。2.乙醇标准溶液制备:取色谱纯无水乙醇适量置于容量瓶中,精密称定,用纯化水稀释至刻度,摇匀,制成1 mg/mL的标准贮备溶液,取上述标准贮备溶液稀释成500-500ug/mL的系列标准溶液。3.样品制备:取医用透明质酸钠凝胶1 g,精密称定,置10 mL顶空瓶中,准确加入纯化水1. 0 mL,混合均匀。另精密量取乙醇标准溶液各1. 0 mL,置10 mL顶空瓶中,准确加人纯化水1. 0 mL,混合均匀。各瓶在80℃的干燥箱中加热30 min,用在同一温度下加热的注射器抽取顶空气体各100uL,在规定的色谱分析条件下,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],待乙醇色谱峰流出后,量取乙醇峰的面积值,作为外标的定量标准。4. 结果计算:医用透明质酸钠凝胶中乙醇的浓度为C=M/m(C医用透明质酸钠凝胶中乙醇的浓度(ug/g), M:标准曲线上查得的医用透明质酸钠凝胶中乙醇残留量(ug) m医用透明质酸钠凝胶的称样量(g) )。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。
药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。 药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。 各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。 按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂 是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如: 苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。 第二类溶剂 是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下: 2-甲氧基乙醇(50ppm)、氯仿(60ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、1,2,3,4-四氢化萘(100ppm)、2-乙氧基乙醇(160ppm)、环丁砜(160ppm)、嘧啶(200ppm)、甲酰胺(220ppm)、正己烷(290ppm)、氯苯(360ppm)、二氧杂环己烷(380ppm)、乙腈(410ppm)、二氯甲烷(600ppm)、乙烯基乙二醇(620ppm)、N,N-二甲基甲酰胺(880ppm)、甲苯(890ppm)、N,N-二甲基乙酰胺(1090ppm)、甲基环己烷(1180ppm)、1,2-二氯乙烯(1870ppm)、二甲苯(2170ppm)、甲醇(3000ppm)、环己烷(3880ppm)、N-甲基吡咯烷酮(4840ppm)、。 第三类溶剂 是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括: 戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。 除上述这三类溶剂外,在药物、辅料和药品生产过程中还常用其他溶剂,如1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙酮、甲基四氢呋喃、石油醚、三氯乙酸、三氟乙酸。这些溶剂尚无基于每日允许剂量的毒理学资料,如需在生产中使用这些溶剂,必须证明其合理性。
药物中常见残留溶剂及其限度 溶剂名称 PDE值 (mg/天) 限度 (%)溶剂名称 PDE值(mg/天) 限度(%)第一类溶剂 (应避免使用) 第三类溶剂(GMP或 其他质量要求限制使用) 苯 0.02 0.0002 乙酸 50.0 0.5 四氯化碳 0.04 0.0004 丙酮 50.0 0.5 1,2-二氯乙烷 0.05 0.0005 甲氧基苯 50.0 0.5 1,1-二氯乙烯 0.08 0.0008 正丁醇 50.0 0.5 1,1,1-三氯乙烷 15.0 0.15 仲丁醇 50.0 0.5 第二类溶剂 (应该限制使用) 乙酸丁酯 50.00.5 乙腈 4.1 0.041 叔丁基甲基醚 50.0 0.5 氯苯 3.6 0.036 异丙基苯 50.0 0.5 氯仿 0.6 0.006 二甲亚砜 50.0 0.5 环己烷 38.8 0.388 乙醇 50.0 0.5 1,2-二氯乙烯 18.7 0.187 乙酸乙酯 50.0 0.5 二氯甲烷 6.0 0.06 乙醚 50.0 0.5 1,2-二甲氧基乙烷 1.0 0.01 甲酸乙酯 50.0 0.5 N,N-二甲氧基乙酰胺10.9 0.109 甲酸 50.0 0.5 N,N-二甲氧基甲酰胺8.8 0.088 正庚烷 50.0 0.5 1,4-二氧六环 3.8 0.038 乙酸异丁酯 50.0 0.5 2-乙氧基乙醇 1.6 0.016 乙酸异丙酯 50.0 0.5 乙二醇 6.2 0.062 乙酸甲酯 50.0 0.5 甲酰胺 2.2 0.022 3-甲基-1-丁醇 50.0 0.5 正己烷 2.9 0.029 丁酮 50.0 0.5 甲醇 30.0 0.3 甲基异丁基酮 50.0 0.5 [t
三氟乙醇[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测!原料药溶剂残留测定,三氟乙醇[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]用什么方法测峰形比较好?
编者按:药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。除上述这三类溶剂外,在药物、辅料和药品生产过程中还常用其他溶剂,如1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙酮、甲基四氢呋喃、石油醚、三氯乙酸、三氟乙酸。这些溶剂尚无基于每日允许剂量的毒理学资料,如需在生产中使用这些溶剂,必须证明其合理性。美、日、欧洲的药典对这一问题的处理不一样,被列入清单的毒性有机溶剂的种类和相应的可接受限度也不相同。虽然国际协调大会(ICH)关于药品中残留溶剂的指导原则在1997年就已生效,但《美国药典》至今尚未完全采纳该指导原则。《美国药典》的残留溶剂检测归在附录中的“有机挥发性杂质”篇,规定只有在生产商指出产品中可能有残留溶剂存在时才进行此检测,而当生产商根据其产品的生产、运输和储藏的相关知识可以保证产品中无某一种溶剂存在,并且保证如果进行此检测的话,产品能符合残留限度要求的时候,就可不进行此检测。同时还认为,装在气密性容器中的物品在运输过程中不受任何溶剂的污染。《美国药典》推荐进行苯、氯仿、二氧杂环己烷、亚甲基氯、三氯乙烯残留量检测。此外,还在一些药品的各论中指定进行环氧乙烷的残留量检测,除非另有规定,环氧乙烷残留量的可接受限度为10ppm。除此以外,《美国药典》不考虑国际协调大会(ICH)指导原则中的其他溶剂。第14版《日本药典》已采用国际协调大会(ICH)的指导原则,将残留溶剂定义为存在于药品中,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行检测,限度符合国际协调大会(ICH)指导原则规定的有机溶剂。《欧洲药典》完全采纳国际协调大会(ICH)关于残留溶剂的指导原则。第4版《欧洲药典》叙述了如何对第一类和第二类溶剂进行鉴别和定量分析的方法,试验方法还适用于第三类溶剂和限度大于1000ppm(0.1%)的第二类溶剂的定量分析。
如题,我们检测中药提取物里面的溶剂残留,比如甲醇,乙醇,乙酸乙酯等,我是用二甲亚砜做提取溶剂,还有更好的选择吗?大家用的提取溶剂是什么级别的,我感觉国产溶剂似乎里面有些杂质峰。还有,检测时间长了,那些经常检测的溶剂,比如甲醇,乙醇,有进样残留,大家有这个问题吗
各位老师,本人是个新手,不知如何做乙醇和异丙醇的残留量测定。本人现有条件如下:1.仪器:GC900A型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](上海科创)2.工作站:国产大连工作站3.色谱柱:极性色谱柱,固定液为聚乙二醇(PEG-20M)的毛细管柱4.检测的为2005年版药典第三类试剂:乙醇和异丙醇药典规定为0.5%我想请教的问题是:1.如何配制对照品溶液?2.操作方法如何?[em04] [em07]
第一步:甲氧基聚乙二醇的合成聚乙二醇在无水二氯甲烷中与金属钠作用生成聚乙二醇钠, 然后与碘甲烷反应即得。一甲氧基聚乙二醉、双端都反应的二一甲氧基聚乙止醇和未反应的聚乙二醇的反应混合物硅胶柱层析色潜提纯可以得到纯净的甲氧基聚乙二醇第二步:甲氧基聚乙二醇丁二酸单醋的合成将甲氧基聚乙二醇(Me-PEG-2000)、丁二酸酐和催化剂加入盛有二氯甲烷的圆底烧瓶中, 磁力搅拌使固体完全溶解后, 室温搅拌反应过夜。反应液分别用盐酸水溶液、氢氧化钠水溶液和甲醇水溶液依次洗涤。有机相经无水MgSO4干燥, 过滤除去干燥剂, 减压蒸除有机溶剂, 残留物以石油醚结晶, 收率90%。第三步:甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺的合成甲氧基聚乙二醇丁二酸单酷先经N一羟基丁二酰亚胺(NHS)活化, 然后缓慢滴加人到二硬脂酰磷脂酰乙醇胺(DSPE)的三氯甲烷中, 加料完毕后继续反应4h, 蒸除溶剂, 浓缩液在乙醚中结晶,硅胶柱层析色谱提纯可以得到自色粉末状固体的。甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺。来源:中国标准物质网
请问一下正己基三乙氧基硅烷,自动进样针的清洗溶剂用什么合适,我用乙醇老是堵????太费针了
有没有高手做过乙醚的残留,用乙醇做溶剂,用DB624的柱子可以做出来吗?两者互相干扰吗?
[align=left][font=宋体]残留溶剂检测方法的选择[/font][/align][align=left][font=宋体]残留溶剂的测定一般采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法,推荐使用毛细管色谱柱[/font]-[font=宋体]顶空进样系统,当然也可以使用普通填充柱,溶液直接进样方法。对不宜采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定的含氮碱性化合物,如[/font]N-[font=宋体]甲基吡咯烷酮等可采用其它方法,如[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法等。[/font][font=宋体]测定残留溶剂应从以下几个方面考虑:确定被测的有机溶剂、选择合适的色谱柱、制备供试品溶液和对照品溶液、选择合适的进样方法和满足检测灵敏度要求的检测器,下面分别进行介绍:[/font] [/align][align=left]1[font=宋体]、确定被测的有机溶剂[/font][font=宋体]根据制备工艺确定被测有机溶剂的范围。通常应对制备工艺过程中使用的二类以上溶剂和重结晶用溶剂,以及根据工艺特点要求的其它溶剂进行残留量的研究。建议对合成最后三步使用的三类溶剂也进行研究,这样能更好地对未知峰进行归属;对制剂过程中使用的有机溶剂也建议考察其残留情况,特别是缓、控释微丸包衣过程使用的有机溶剂更应引起注意。残留溶剂的限度要求同[/font]ICH[font=宋体]的规定。[/font] [/align][align=left]2[font=宋体]、选择合适的色谱柱[/font][font=宋体]按照相似相溶的原理选择色谱柱。毛细管柱有极性柱、非极性柱、弱极性柱和中等极性柱。填充柱有高分子多孔小球或涂渍适宜固定液的填充柱。[/font] [/align][align=left][font=宋体]测定含氮的碱性有机溶剂时,由于普通[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]的不锈钢管路、进样器衬管等对有机胺等含氮的碱性化合物具有较强的吸附作用,致使其检出的灵敏度降低。通常采用弱极性色谱柱或经碱处理过的色谱柱分析含氮的碱性有机溶剂,如果采用胺分析专用柱进行分析,则效果更好。[/font] [/align][align=left]3[font=宋体]、供试品和对照品的制备[/font][font=宋体]顶空进样方法通常以水为溶剂,对于非水溶性的药物,可采用[/font]DMF[font=宋体]、[/font]DMSO[font=宋体]或其他适宜溶剂。溶液直接进样方法用水或合适的溶剂溶解样品。[/font][font=宋体]制备供试品的溶剂的选择应兼顾供试品和被测有机溶剂的溶解度,且所用溶剂应不干扰被测有机溶剂的测定。水是首选溶剂,特别是顶空进样系统。因为水中不含有机溶剂,故干扰较少,且在[/font]FID[font=宋体]检测器上,以水为溶剂时,各残留溶剂的灵敏度最高。当药物不溶于水时,可加入适当的酸或碱以增加药物的溶解度,最好选用不挥发的酸或碱。以[/font]DMSO[font=宋体]等为溶剂时,可加入一定量的水以增加检测的灵敏度,或用盐析的方法增加灵敏度。测定含氮的碱性溶剂时,供试品溶液应不呈酸性,以免被测物与酸反应后不易汽化。[/font] [/align][align=left][font=宋体]对照品的制备方法应与供试品的制备方法相同。在申报资料中发现对照品(溶液)为直接进样,供试品则为固体直接顶空进样,供试品和对照品不但制备方法不同,而且进样方法和进样量也不同,无法进行比较。提请申报单位注意。[/font] [/align][align=left]4[font=宋体]、供试品溶液和对照品溶液浓度的确定配制供试品溶液的浓度应满足定量测定的需要,一般供试品取样量在[/font]0.1[font=宋体]~[/font]1g[font=宋体]之间。限度检查时对照品溶液的浓度可按规定的限度配制,定量测定时按实际残留量配制,浓度相差最好以不超过[/font]2[font=宋体]倍为宜。[/font] [/align][align=left]5[font=宋体]、检测器的选择[/font][font=宋体]一般选用[/font]FID[font=宋体]检测器,对含卤素的有机溶剂如氯仿等,采用[/font]ECD[font=宋体]检测器可得到更高的灵敏度。通常可根据药物溶剂的残留情况选择合适的检查方法。当需要检查的有机溶剂数量不多,且极性差异较小时,可选择毛细管色谱柱[/font]-[font=宋体]顶空进样[/font]-[font=宋体]等温法。当需要检查的有机溶剂数量较多,且极性、沸点差异较大时,可选择毛细管色谱柱[/font]-[font=宋体]顶空进样[/font]-[font=宋体]程序升温法;也可选择填充柱或适宜极性的毛细管柱直接进样法。[/font][font=宋体]限度检查(一类、部分二类溶剂)时采用内标法或外标法;定量测定(含量超过[/font]0.1%[font=宋体]的二类或需要定量控制的三类溶剂)时采用内标法或标准加入法。[/font] [/align][align=left][font=宋体]顶空进样法还要对顶空温度和顶空时间进行选择。[/font][font=宋体]顶空温度应根据溶解供试品溶剂的特性及供试品中残留溶剂的沸点选择。以水为溶剂及测定低沸点残留溶剂时,顶空温度不宜超过[/font]85[font=宋体]℃[/font][font=宋体];测定沸点较高的残留溶剂时,通常选择较高的顶空温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物干扰测定结果。以[/font]DMSO[font=宋体]为溶剂时,顶空温度不宜超过[/font]115[font=宋体]℃[/font][font=宋体]。例如,在申报资料中发现,以水为溶剂,顶空温度为[/font]100[font=宋体]℃[/font][font=宋体],柱温[/font]60[font=宋体]℃[/font][font=宋体],结果高浓度的乙腈比低浓度的二氯甲烷的峰面积还小,原因是顶空温度太高,大量的水被蒸发(或浓度被稀释),随着水蒸汽的凝结,在水中溶解度大的乙腈的灵敏度下降,产生了与事实不符的实验结果。[/font] [/align][align=left][font=宋体]顶空时间是要确保供试品溶液的气[/font]-[font=宋体]液两相达到平衡,一般通过测定顶空时间与顶空气体浓度的浓度[/font]—[font=宋体]时间曲线来确定。顶空时间不宜过长,一般为[/font]30[font=宋体]~[/font]45[font=宋体]分钟,如果超过[/font]60min[font=宋体],可能引起顶空瓶的气密性变差[/font],[font=宋体]导致定量的准确性降低。如果平衡时间选择[/font]10[font=宋体]分钟,就不能保证气[/font]-[font=宋体]液两相达到平衡。注意对照品溶液与供试品溶液必须使用相同的顶空条件。[/font][font=宋体]甲酰胺、[/font]2[font=宋体]-甲氧基乙醇、[/font]2[font=宋体]-乙氧基乙醇、乙二醇、[/font]N-[font=宋体]甲基吡咯烷酮等不适宜用顶空法测定。[/font][/align]
各位有机残留分析方法验证中用到的有机溶剂(如甲醇、乙醇、二氯甲烷等)是什么级别的?中国药品生物制品检定所提供这些溶剂的对照品?
[color=#444444]我最近在做一个粉末原料的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]顶空进样残留溶剂方法验证。残留溶剂为甲醇、乙醇。残留溶剂限度按照内部质量标准的限度为甲醇:1% 乙醇:2%。[/color][color=#444444]我初步确定顶空瓶的装样量为5ml,供试品浓度为40mg/ml。[/color][color=#444444]这样做出来,我的定量限浓度为100%标准溶液的1.25%。[/color][color=#444444]接下来我要做线性,我想问的是线性要求最低浓度为定量限浓度。但是我的定量限浓度太低了,如果把定量限包括进去,可能线性结果不太好。[/color][color=#444444]不知道要怎么改进这个方案。[/color][color=#444444]还有,把定量限控制在标准溶液浓度的多少倍比较合适,有没有这方面的要求?[/color][color=#444444]还有顶空进样,同一个样品可以多次进样吗?做线性的话,每个浓度的样品进多少个平行样?[/color][color=#444444]谢谢![/color]
采用HP-5柱,顶空方法,水作溶剂,加热85摄氏度,平衡30min,进样口温度200摄氏度,分流比10:1,程序升温:40摄氏度保持5min,5摄氏度每分钟升温至150。FID检测器。混合液:DMF浓度160mg/L,乙醇800mg/mL.DMF未检测出,乙醇出峰,响应非常大。查阅文献,方法均相似,分离效果和相应均很好,但为何我的实验DMF未出峰?如何调整使二者均能出峰,且相应较好?
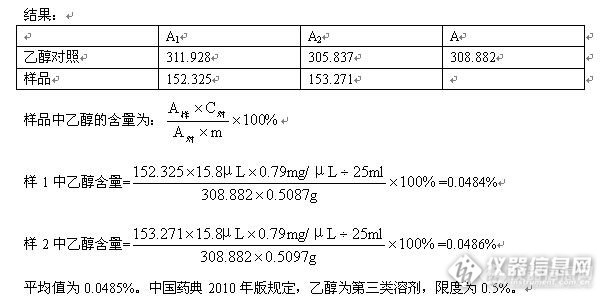
大豆磷脂是一种混合磷脂,它是由磷脂酰胆碱(卵磷脂,简称PC,高等级为PPC)、磷脂酰乙醇胺(脑磷脂,简称PE)、磷脂酰肌醇(肌醇磷脂,简称PI)、磷脂酰丝胺酸(丝胺酸磷脂,简称PS)等成分组成,其中最典型的是前三种。磷脂是人体细胞(细胞膜、核膜、质体膜)的基本成分,并对神经、生殖、激素等功有重要关系,具有很高营养价值和医用价值。现代人生活节奏紧张,磷脂营养大量流失,因此补充完整磷脂(PC、PE、PI…)对现代人而言是绝对必要。鉴于大豆磷脂类保健品是一种功能性的健康食品,虽然不是立即见效,但有著全面、长远、稳定的效果,同时又没有药物的副作用,医学家们也开始重视卵磷脂在预防疾病发生方面的积极作用。背景介绍:对于我们的项目来讲,很多原料的检验是要用到GC的。GC到位后,很多以前自己无法解决的检验项目,现在都是自己做了。六月初到货了一批大豆磷脂,就需要检验其中乙醇的溶剂残留。大豆磷脂为中国药典2010年版已经收载的药用辅料,在正文第二部分的1183-1184页上。我们的检验,就是按照这个药典标准进行的。实验步骤:对照品溶液的制备:先向25ml的容量瓶中加入适量的水,然后用微量注射器精密量取15.8μL乙醇对照品(密度按0.79g/ml计,15.8μL的乙醇的质量为12.48mg),然后用水稀释并定容至刻度。精密量取5ml置顶空瓶中,密封。供试液制备:取供试品约0.5g,精密称定,置顶空瓶中,精密加入水5ml使其分散,密封。色谱条件:月旭WEl-PEG20M气相色谱柱,30m*0.32mm*0.25μm(Cat. NO:01918-32001;Ser. NO:GC201311
一、概述药物中的残留溶剂系指在原料药或辅料的生产中,以及制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。按有机溶剂的毒性和对环境的危害,ICH将有机溶剂分为避免使用、限制使用、低毒和毒性依据尚不足四种情况。因残留溶剂会影响产品的安全性,故需对其进行研究。二、残留溶剂检查方法的选择残留溶剂的测定一般采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,推荐使用毛细管色谱柱-顶空进样系统,当然也可以使用普通填充柱,溶液直接进样方法。对不宜采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定的含氮碱性化合物,如N-甲基吡咯烷酮等可采用其它方法,如[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法等。测定残留溶剂应从以下几个方面考虑:确定被测的有机溶剂、选择合适的色谱柱、制备供试品溶液和对照品溶液、选择合适的进样方法和满足检测灵敏度要求的检测器,下面分别进行介绍:1、确定被测的有机溶剂根据制备工艺确定被测有机溶剂的范围。通常应对制备工艺过程中使用的二类以上溶剂和重结晶用溶剂,以及根据工艺特点要求的其它溶剂进行残留量的研究。建议对合成最后三步使用的三类溶剂也进行研究,这样能更好地对未知峰进行归属;对制剂过程中使用的有机溶剂也建议考察其残留情况,特别是缓、控释微丸包衣过程使用的有机溶剂更应引起注意。残留溶剂的限度要求同ICH的规定。2、选择合适的色谱柱按照相似相溶的原理选择色谱柱。毛细管柱有极性柱、非极性柱、弱极性柱和中等极性柱。填充柱有高分子多孔小球或涂渍适宜固定液的填充柱。测定含氮的碱性有机溶剂时,由于普通[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的不锈钢管路、进样器衬管等对有机胺等含氮的碱性化合物具有较强的吸附作用,致使其检出的灵敏度降低。通常采用弱极性色谱柱或经碱处理过的色谱柱分析含氮的碱性有机溶剂,如果采用胺分析专用柱进行分析,则效果更好。3、供试品和对照品的制备顶空进样方法通常以水为溶剂,对于非水溶性的药物,可采用DMF、DMSO或其他适宜溶剂。溶液直接进样方法用水或合适的溶剂溶解样品。制备供试品的溶剂的选择应兼顾供试品和被测有机溶剂的溶解度,且所用溶剂应不干扰被测有机溶剂的测定。水是首选溶剂,特别是顶空进样系统。因为水中不含有机溶剂,故干扰较少,且在FID检测器上,以水为溶剂时,各残留溶剂的灵敏度最高。当药物不溶于水时,可加入适当的酸或碱以增加药物的溶解度,最好选用不挥发的酸或碱。以DMSO等为溶剂时,可加入一定量的水以增加检测的灵敏度,或用盐析的方法增加灵敏度。测定含氮的碱性溶剂时,供试品溶液应不呈酸性,以免被测物与酸反应后不易汽化。对照品的制备方法应与供试品的制备方法相同。在申报资料中发现对照品(溶液)为直接进样,供试品则为固体直接顶空进样,供试品和对照品不但制备方法不同,而且进样方法和进样量也不同,无法进行比较。提请申报单位注意。4、供试品溶液和对照品溶液浓度的确定配制供试品溶液的浓度应满足定量测定的需要,一般供试品取样量在0.1~1g之间。限度检查时对照品溶液的浓度可按规定的限度配制,定量测定时按实际残留量配制,浓度相差最好以不超过2倍为宜。5、检测器的选择一般选用FID检测器,对含卤素的有机溶剂如氯仿等,采用ECD检测器可得到更高的灵敏度。通常可根据药物溶剂的残留情况选择合适的检查方法。当需要检查的有机溶剂数量不多,且极性差异较小时,可选择毛细管色谱柱-顶空进样-等温法。当需要检查的有机溶剂数量较多,且极性、沸点差异较大时,可选择毛细管色谱柱-顶空进样-程序升温法;也可选择填充柱或适宜极性的毛细管柱直接进样法。限度检查(一类、部分二类溶剂)时采用内标法或外标法;定量测定(含量超过0.1%的二类或需要定量控制的三类溶剂)时采用内标法或标准加入法。顶空进样法还要对顶空温度和顶空时间进行选择。顶空温度应根据溶解供试品溶剂的特性及供试品中残留溶剂的沸点选择。以水为溶剂及测定低沸点残留溶剂时,顶空温度不宜超过85℃;测定沸点较高的残留溶剂时,通常选择较高的顶空温度;但此时应兼顾供试品的热分解特性,尽量避免供试品产生的挥发性热分解产物干扰测定结果。以DMSO为溶剂时,顶空温度不宜超过115℃。例如,在申报资料中发现,以水为溶剂,顶空温度为100℃,柱温60℃,结果高浓度的乙腈比低浓度的二氯甲烷的峰面积还小,原因是顶空温度太高,大量的水被蒸发(或浓度被稀释),随着水蒸汽的凝结,在水中溶解度大的乙腈的灵敏度下降,产生了与事实不符的实验结果。顶空时间是要确保供试品溶液的气-液两相达到平衡,一般通过测定顶空时间与顶空气体浓度的浓度—时间曲线来确定。顶空时间不宜过长,一般为30~45分钟,如果超过60min,可能引起顶空瓶的气密性变差,导致定量的准确性降低。如果平衡时间选择10分钟,就不能保证气-液两相达到平衡。注意对照品溶液与供试品溶液必须使用相同的顶空条件。甲酰胺、2-甲氧基乙醇、2-乙氧基乙醇、乙二醇、N-甲基吡咯烷酮等不适宜用顶空法测定。
本文简要介绍了软包装领域中溶剂残留量超标所引发的社会问题,结合成因及对策,深入介绍软包行业的专用检测设备——由北京兰德梅克包装器材有限公司生产的2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的技术应用。一、食品包装类溶剂残留量超标所引发的社会问题 去年,随着央视对“甘肃食品袋毒袋”事件的曝光,社会各界对存在危及人体安全隐患——食品医药类复合包装用薄膜的“溶剂残留量超标”问题予以了充分关注……(载:国家质量技术监督局GB/T200中明文规定:包装食品类溶剂残留总量应≤10mg/m² ,其中苯类溶剂残留量≤3mg/m² )意即:如果我们使用的各种包装袋中溶剂残留量超过此标准,那么残留溶剂中的甲苯、二甲苯、丁酮、异丙醇、乙酸丁酯、乙酸乙酯等物质将像无形杀手一样对人体安全产生巨大威胁! 所以,“控制溶剂残留量,保证食品安全”的社会责任已不容置疑地将我们软包装企业推入了舆论的焦点。那么,究竟何种原因导致了溶剂残留量超标?该如何进行生产的过程控制?专用的“2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]”如何检测溶剂残留量?检测实验中应注意哪些问题?对已超标的复合薄膜又将做何处理?以下将作详细说明:二、溶剂残留异味的成因及对策 对软包装厂而言,造成溶剂残留超标(有异味)的因素有很多,诸如:选择基材不当、生产工艺不规范,设备过于简陋老化,生产各环节没有进行必需的检测控制 等等,都与超标(异味)有着直接或间接的影响。拿原料基材来讲:油墨的品质、油墨的释放性以及涂墨的厚度,胶粘剂的品质、涂布量等,有机溶剂的纯度和配比 量以及与基材的相符性,都将影响溶剂的残留量。另外,软包生产设备过于简陋老化导致加热温度不合适,烘干风量不足,或无明晰之作业指导书,员工操作不规范,开机印刷速度不达标,添加剂不足,涂布不均匀、成品包装用材料较随意等等,都是造成其超标的主要因素。 特别值得一提的是:为数不少的软包企业在整个生产过程当中,完全省略了“品质检测”这一重要控制环节,根本没有配置专业用于检测溶剂残留异味及纯度的专用检测设备,就“包装袋异 味”的检测,仅凭经验依靠人的嗅觉进行简单判定,极大程度地增加了溶剂超标的发生机率,给使用企业及社会将造成重大危害。曾经有家企业在9月初,将已产成 的有严重异味的口罩制袋,拿去北京兰德梅克包装器材有限公司用2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进行检测,结果发现:苯类含量已达7mg/m² !严重超标造成异味!给该企业造成的直接经济损失达上百万(还不算将这些已超标复合膜的重新处理费用)!试想:如果该企业提早注意,加强重要过程的品质控制,是可以避免这种经济损失的。 综合以上分析,可以定论的是:合理的复合层结构,合理的原材料采购和使用,正确的生产工艺,配置专业的检测设备,执行严格标准的检测手法,生产合格安全的复合包装是完全可以的。下文将针对软包装检测,重点介绍2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的应用。三、软包装领域专业检测设备——2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的应用:2061C型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]设计基于色谱分析与氢火焰离子化检测原理。由于氢火焰离子化检测器对有机化合物特别敏感,最小检测器可达10-9克,而对无机化合物无响应或响应很小。FID响应特性属于质量检测器,因此对温度、压力、流量等操作条件极不敏感,具有其他常用检测器无法比拟的操作特性,是目前[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]必配的检测器。值得指出的是,氢火焰离子化检测器也是目前[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]常用检测中唯一可以进水样的检测器。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的工作原理:以气体作为流动相、固体作固定相的色谱法。所需要分离的组分,分配在流动相和固定相两相中,色谱过程就是样品组分在通过色谱柱时,不断发生反复的吸附和脱附,或溶解和解析。由于各组分的吸附能或分配系数不同,沿载气方向移动速度不同,相继从柱中洗脱出的时间则不同,从而使组分得到分离。从柱中流出的组分进入检测器,将化学信号转变成电信号,以电压的形式被记录下来,得出各组分的定性和定量信息。 2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]是一种专业用于准确、快速检测印刷包装品溶剂残留含量及检测溶剂纯度的高精度智能化检测系统,具有定性定量分析速度快、对比表现直观、数据精确可靠等系列特点: 1、不仅能直接显示出包装膜的各残留溶剂名称,如:乙醇、异丙醇、乙酸乙酯、乙酸丁酯、丁酮、苯类(甲苯,二甲苯),而且无须核算和计量可直接显示出包装膜每平方米多少毫克(mg/m² )的溶剂残留含量,结果直观、方便、数据精确可靠。 2、除可检测包装膜的溶剂残留含量,还可检测所使用溶剂的纯度,鉴别无味溶剂的成分特性,如乙醇、异丙醇、乙酸乙酯、乙酸丁酯、丁酮、苯类(甲苯,二甲苯)等溶剂,经过色谱检测,均可直接快速测得各含量及杂质等未知物,此举可有效防止溶剂的掺杂使假,杜绝不合格品的使用。 通过2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]的检测,可为软包装企业有效调整生产工艺,控制溶剂,为防止溶剂残留超标提供准确方向,例如苯类超标,可立即判定印刷过程需要调整之工艺 参数,提高溶剂的挥发效率;若乙脂超标,则可判断干复工艺存在的问题,需要调整;对印刷半成品进行检测,则可有效判定印刷过程溶剂量指标,为生产质量控制 提供指引,对复合过程进行检测,则可有效判别复合过程该溶剂的指标。 所以通过2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]上述检测说明,来判断该包装使用复合薄膜的溶剂残留量是否超标,是否需要重新开发生产工艺,对所采用的基材是否恰当等提供理论与技术支持,可以从根本上控制残留量超标的产生。值得一提的是:取样代表性、复合前的首检及熟化后的检验都是使用2061C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]需重点考虑的地方。[color=red]【由于该附件或图片违规,已被版主删除】[/color][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=18862]软包装溶剂残留异味及溶剂纯度检测[/url]
最近做有机溶剂残留,同时测6种有机溶剂,均稀释溶解在水中,分别为甲醇、乙醇、丙酮、二氯甲烷、三氯甲烷、四氢呋喃。顶空进空白水样,乙醇峰位处有干扰,调整色谱条件,干扰仍无法消除。换用娃哈哈纯净水,也有相似的干扰存在。且峰面积差不多。目前在做这种有机溶剂的检测限和定量限,干扰峰的信噪比将近8,无法测乙醇的信噪比,有想法将空白水样的干扰峰去除,来做定限量。(Agilent 7890带顶空进样器)软件中可以自动扣除空白。不知道是否可以这样做
使用的乙醇作溶剂,就要测苯吗?最近有关有经验的人说,在合成中使用了乙醇、丙酮,那么溶剂残留的测定以及方法学验证时就要测苯,我觉得很疑惑,不知道待检溶剂怎么确定才好,请各位大师指点一下我的迷津?







 我要推广仪器
我要推广仪器
 下载APP
下载APP