现在在做乙氧基喹啉的残留检测,手头的方法有:高效液相色谱C18柱UV检测器,高效液相色谱C18柱荧光检测器,还有GB/T17814-1999的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]FID检测,请问各路大侠,他们有什么差别?对检测的结果影响大吗?检测的极限差的多吗?
徐建强(南京气象学院环境科学系,南京 210044)分光光度法是获得物质光吸收特性及定性、定量分析的重要手段,在冶金、地质、生物、医学、农业、环境监测、食品卫生等部门得到极其广泛的应用。分光光度法的发展不仅依赖于电子学、激光和计算机技术的发展和应用,而且还依赖于高灵敏度、高选择性有机试剂的合成和应用。喹啉类试剂作为光度分析的有机试剂,可以分为两类,一类是喹啉及其衍生物,如8-羟喹啉、8-巯基喹啉、8-氨基喹啉等,这类试剂可用作金属离子光度分析的显色剂,具有一定的灵敏度和选择性 另一类是喹啉偶氮化合物,喹啉偶氮化合物作为一大类显色剂已有多种试剂被合成和研究。其中8-氨基喹啉的8位偶氮衍生物前苏联学者研究得较早 我国的李亚文等也进行了系统深入的研究,近年来不断有一系列新的衍生物合成,这些化合物因其特有的灵敏度和选择性而备受化学工作者的关注。自从1955年程广禄等[3]首次提出PAN(即1-(2-吡啶偶氮)-2-萘酚)作为分析试剂后,越来越多的偶氮试剂相继被化学工作者合成并且用于光度分析研究。实践证明,偶氮化合物(azo-compounds)具有性质稳定、显色反应灵敏度高、选择性好、对比度大等优点,仍然是目前应用最广泛的一类显色剂。本文选用6-甲氧基-8-氨基喹啉作为原料,首先对其进行重氮化,得到重氮盐,然后与间苯二酚进行偶联反应,合成了新有机试\剂:4-(6-甲氧基-8-喹啉偶氮)-间苯二酚(简称MQAR)。对其进行了结构分析和鉴定。实验研究表明,MQAR能与Co、Cu、Fe、Ni等金属离子发生灵敏的显色反应,该试剂可用于试样中微量金属离子的测定(另文介绍),方法简便、快速、准确可靠,是一种比较理想的新有机试剂。1 合成方法1.1 试剂和仪器设备6-甲氧基-8-氨基喹啉(由工业品提纯所得),亚硝酸钠(AR),间苯二酚(AR),N,N-二甲基甲酰胺(AR),浓硫酸(AR),甲酸(AR)。中量有机化学制备仪、真空干燥箱、差热分析仪、Perkin-Elmer元素分析仪、IR-408型红外分光光度计、756MC型紫外-可见分光光度计、BRUKERARX300M核磁共振谱仪。1.2 合 成1.2.1 合成线路MQAR的合成线路如图1所示。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221937_18814_1634962_3.gif[/img]1.2.2 合成步骤(1)重氮化 在250mL三口烧瓶中,加入8.7g6-甲氧基-8-氨基喹啉,10mL甲酸,同时加入由15mL浓硫酸和10mL水配制而成的溶液,搅拌溶解,置于冰浴中冷却。在0~5℃时边搅拌边滴加3.5g亚硝酸钠与10mL水配制而成的溶液,控制在1h内加完,使反应充分完全,最后得到深红色的重氮盐溶液,用于下一步的反应。(2)偶联 取5.5g间苯二酚溶于75mL无水乙醇中,置于冰浴中冷却。在0~5℃时,边搅拌边加入上述重氮盐溶液,控制重氮盐溶液于1h内加完,继续搅拌2h,静置过夜得砖红色沉淀,抽滤,依次用水、无水乙醇洗涤,抽干。干燥后得棕黄色粗品。(3)精制 将偶联得到的粗品用N,N-二甲基甲酰胺重结晶两次,于真空干燥箱干燥,得到深红色的MQAR纯品。经测定产品熔点为194℃。2 结构分析2.1 薄层色谱分析(1)试剂和仪器设备 展开剂正丁醇∶无水乙醇∶2molL-1氨水=3∶1∶1 支持剂硅胶HF254+0.3%CMC 8×10cm2薄层板(自制) 层析缸。(2)结果和讨论 在不同极性的溶剂体系中进行展开,在薄层色谱板上只发现一个斑点,在紫外灯光下未发现其他斑点,(1)中所列展开剂效果最好,比移值Rf=0.66(图2)。结果表明,产品中只含有一种物质。2.2 元素分析用Perkin-Elmer元素分析仪对产品进行了元素分析,产品MQAR元素分析结果与理论计算值基本一致。2.3 紫外-可见吸收光谱分析用756MC型紫外-可见分光光度计测得2×10-5molL-1MQAR的10%DMF水溶液(pH=8.3时)的紫外-可见吸收光谱(图3)。Kmax=450nm,E=2.98×104Lmol-1cm-1。结果表明,产品分子是一个大的共轭体系。2.4 红外吸收光谱分析用IR-408型红外分光光度计对产品MQAR进行了红外吸收光谱分析(KBr压片法),产品的红外光谱解析结果见表1。解析结果表明,产品分子中含有酚羟基、芳环、偶氮基等官能团,还具有1,2,4-三取代苯结构。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221954_18815_1634962_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18816_1634962_3.gif[/img]2.5 核磁共振谱分析用BRUKERARX300M核磁共振谱仪(DMSO-d6溶剂,TMS内标)对产品进行核磁共振谱分析,得到化学位移、峰面积等(表2)。解析结果表明,产品分子中除了羟基质子以外,还含有甲氧基质子以及苯环和杂环质子。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18817_1634962_3.gif[/img]2.6 产品的结构根据实验条件以及结构鉴定和分析,可以确定产品4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的分子结构为[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221956_18818_1634962_3.gif[/img]3 结 论研究了新有机试剂4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的合成方法、实验条件和精制方法,通过薄层色谱法、元素分析、紫外-可见吸收光谱法、红外吸收光谱法和核磁共振谱法等分析手段,对合成的产品进行了分析和结构鉴定,实验结果确证合成了4-(6-甲氧基-8-喹啉偶氮)-间苯二酚纯品。
谁知道微量乙氧基喹啉怎么测定,我好着急呀,哪为仁兄可以告诉我吗
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=167283]水果中乙氧基喹残留量的检验方法[/url]
大家有没有喹啉铜残留检测的方法?
求助:苄嘧磺隆和二氯喹啉酸混剂在水稻上的残留检测参考资料,还有苄嘧磺隆和二氯喹啉酸的最新MRL值,谢谢!我是新手,在中国知网和重庆维普上查了一些,但感觉不够,所以想请各位高手指点迷经,谢谢了!
编者按:药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。除上述这三类溶剂外,在药物、辅料和药品生产过程中还常用其他溶剂,如1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙酮、甲基四氢呋喃、石油醚、三氯乙酸、三氟乙酸。这些溶剂尚无基于每日允许剂量的毒理学资料,如需在生产中使用这些溶剂,必须证明其合理性。美、日、欧洲的药典对这一问题的处理不一样,被列入清单的毒性有机溶剂的种类和相应的可接受限度也不相同。虽然国际协调大会(ICH)关于药品中残留溶剂的指导原则在1997年就已生效,但《美国药典》至今尚未完全采纳该指导原则。《美国药典》的残留溶剂检测归在附录中的“有机挥发性杂质”篇,规定只有在生产商指出产品中可能有残留溶剂存在时才进行此检测,而当生产商根据其产品的生产、运输和储藏的相关知识可以保证产品中无某一种溶剂存在,并且保证如果进行此检测的话,产品能符合残留限度要求的时候,就可不进行此检测。同时还认为,装在气密性容器中的物品在运输过程中不受任何溶剂的污染。《美国药典》推荐进行苯、氯仿、二氧杂环己烷、亚甲基氯、三氯乙烯残留量检测。此外,还在一些药品的各论中指定进行环氧乙烷的残留量检测,除非另有规定,环氧乙烷残留量的可接受限度为10ppm。除此以外,《美国药典》不考虑国际协调大会(ICH)指导原则中的其他溶剂。第14版《日本药典》已采用国际协调大会(ICH)的指导原则,将残留溶剂定义为存在于药品中,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行检测,限度符合国际协调大会(ICH)指导原则规定的有机溶剂。《欧洲药典》完全采纳国际协调大会(ICH)关于残留溶剂的指导原则。第4版《欧洲药典》叙述了如何对第一类和第二类溶剂进行鉴别和定量分析的方法,试验方法还适用于第三类溶剂和限度大于1000ppm(0.1%)的第二类溶剂的定量分析。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。
2014年4月8日,加拿大卫生部发布PMRL2014-15、PMRL2014-14号通报,有害生物管理局提议修订二氯喹啉酸(Quinclorac)和吡噻菌胺(Penthiopyrad)分别在油菜等作物和芥末种子中的最大残留限量,具体修订信息如下: 通用名MRL(ppm)食品类别二氯喹啉酸1.5油菜或作物亚组20A吡噻菌胺1.5芥末的种子(调味品类)
本人菜鸟在做溶剂残留VOCs比对试验中,26种物质,混标溶液中有5种物质,分高浓度与低浓度两套标液,5种物质分别是甲醇、2-乙氧基乙醇、苯、环己酮、丁二酸二甲酯(按保留时间先后排列),其中甲醇、苯、丁二酸二甲酯均未出现异常,均为正偏离;但是2-乙氧基乙醇检测结果显示,高低两个浓度结果是所有实验室中的极小值,低浓度结果是离群,而环己酮的两个浓度结果为可疑,正偏离方向,求大神指教,应该如何着手分析,试验?
谁有环氧乙烷残留量分析—比色分析法德操作步骤和作业指导书啊?
日本对于越南的虾检查力度很大,对于中国的虾检测很少。现在日本开始修订了乙氧喹啉在大米等产品中的残留标准,将大部分产品中乙氧喹啉的残留限量从0.05ppm调低为一律标准0.01ppm,其中包括大米、小麦、大麦和茶叶。谈谈你对乙氧喹啉的看法。

[size=16px] 农药残留检测仪如何分析生菜中农残残留 农药残留检测仪是一种用于快速检测农产品中农药残留量的仪器。生菜作为蔬菜的一种,其农药残留情况也需要进行检测。以下是使用农药残留检测仪分析生菜中农残残留的步骤: 准备试剂和样品:根据检测的需要准备相应的试剂和生菜样品,并对样品进行预处理,如洗净、晾干等。 加样和反应:将生菜样品和试剂加入到农药残留检测仪中,按照设定的程序进行反应。 检测和分析:在规定的时间内,对反应产生的光学信号进行检测和分析,得出检测结果。 结果报告:根据检测结果,生成报告并进行分析解释。 在分析检测结果时,需要考虑以下几个因素: 检测项目的种类和数量:需要考虑检测项目中涵盖的农药种类和数量,以及这些农药对人类健康的危害程度。 检测结果的准确性和可靠性:需要确保检测结果的准确性和可靠性,以保证对生菜中农药残留量的正确判断。 检测结果的标准和参考值:需要参考相应的标准和参考值对检测结果进行分析和评估,以确定生菜是否符合食品安全标准。 生菜样品的差异:需要考虑不同品种、产地和生长环境等因素对生菜样品的影响,以及这些差异对检测结果的影响。 总之,使用农药残留检测仪可以快速、准确地检测生菜中的农药残留量,评估和分析生菜的安全性和质量,保护消费者的健康和权益。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2023/11/202311271017395378_3295_6098850_3.jpg!w690x690.jpg[/img][/size]
顶空进样一般是不分流进样吗?我做溶剂残留,(初学),不知改了什么条件,不出峰了.柱子是SE-30,样品中有苯,甲苯等,能帮我分析下原因吗?多谢
整理了一些用气象色谱分析的应用资料,供大家分享! 今后会陆续整理上传应用资料的,请大家关注哦!1. 10类农药残留的分析方法2. 氨纶生产过程中胺的分析3. 包装食品中溶剂残留的分析4. 分析薄膜包衣片中的二氯甲烷5. 顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析护肤品中芳香成分(甲基吡嗪)6. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析溶剂纯度(氯仿)7. 挥发性有机酸的分析(氯乙酸,[font=Arial]三氯乙酸,[font=MingLiU]溴乙酸,[font=MingLiU]二氯乙酸,二溴乙酸)8. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析饮用水中残留农药成分9. 牙膏增白剂中有毒气体的分析 10. 液相色谱法分析乳酸钠11. 在线分析工业中痕量废气12. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析烟草中苯、异丁酯、正丁醇、丙二醇甲醚等挥发性有机物13. 永久性气体分析系统分析标准气样的方法14. 对变压油中气体的分析15. 热裂解[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法对聚乙烯的分析16. 热裂解[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法对尼龙66的分析17. 食用色素中残留溶剂的分析方法18. 氨基酸分析中的两种衍生化方法19. 食用包装材料中残留溶剂的分析方法20. 一般残留溶剂的分析方法 (丙酮,四氢呋喃,乙酸乙酯,甲醇,二氯甲烷,苯,乙腈,氯仿,甲苯)21. 药物中残留溶剂的分析方法2[/font][/font][/font][font=MingLiU] (二氯甲烷[/font][font=宋体],[/font][font=MingLiU]氯仿[/font][font=宋体],[/font][font=MingLiU]三氯乙烯[/font][font=宋体],[/font][font=MingLiU]1,4-[/font][font=MingLiU]二氧六环[/font])22. 药物中残留溶剂的分析方法[font=MingLiU] (甲醇[/font][size=3][font=Times New Roman] [/font][/size][font=Times New Roman][/font][font=宋体],[/font][font=MingLiU]丙酮[/font][font=宋体],[/font][font=MingLiU]乙酸乙酯[/font][font=宋体],[/font][font=MingLiU]1.4-[/font][font=MingLiU]二氧六环[/font][font=宋体],[/font][font=MingLiU]吡啶[/font][font=宋体],[/font][font=MingLiU]二甲基甲酰胺[/font][color=black][font=MingLiU])23. 应用PDECD检测器分析农药类物质24. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测试酒样中31种混标25. 顶空法检测制糖原材料中溶剂残留(甲醇、乙醇)[/font][/color]
[align=center][b][size=32px]溶剂残留[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]分析方法操作要点与注意事项[/size][/b][/align]对于溶剂残留的检测首先要明确可能会有的残留溶剂,这些溶剂的分类以及检测要求。确定可能的残留溶剂:残留溶剂的检测首先要明确有可能会有哪些溶剂残留,也就是溶剂有可能在哪个环节带入,从原料、制备工艺、处方各个方面都需要考虑可能的残留溶剂,再根据残留溶剂的类型确定残留溶剂的残留量限值。[b][size=20px][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]分析方法注意事项[/size][/b]对于溶剂残留检测通常用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]来进行检测,在某些情况也会用到高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]等,而残留溶剂的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测方法的开发相对简单,适用范围广,实际运用中不同的溶剂需要再调整,小编根据实际的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]使用经验需要注意的点进行总结:1、进样方式推荐使用的是顶空进样和溶液进样,溶液进样有很大的溶剂峰,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]一般都使用低沸点的溶剂,这样会对低沸点的溶剂残留检测造成很大的干扰,所以一般溶液进样用于高沸点的溶剂检测,顶空进样用于低沸点的溶剂检测。顶空进样时,通常用水作溶剂,水溶液中溶剂容易挥发到顶空气体中,增加检测灵敏度;对于一些极性组分,可以利用盐析作用来增加挥发性;如果非水溶性药品,可使用N,N-二甲基甲酰胺、二甲基亚砜等为溶剂。简而言之就是顶空进样中,应尽量让有机溶剂从样品中挥发出来,才能使检测的灵敏度和准确度增加。顶空温度的选择的原则是必须让待测组分都能从样品中挥发到顶空中,兼顾样品中物质的热稳定性,以免带入干扰物质。平衡时间也是和温度相对应的,温度越高,平衡时间越短;一般情况使用药典建议的就可以。2、色谱柱的选择顶空进样一般选用毛细管色谱柱,溶液进样选择合适的分流比也可以选择毛细管色谱柱,毛细管柱分离效率高,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱固定液的选择根据“相似相溶”的原理,以待分析的化合物的极性选择合适的色谱柱,相同极性的色谱柱之间可替换,小编以前实验室常备的是HP-WAX和HP-5的色谱柱,基本满足大部分的分析需求,常用的色谱柱见下表:[align=center][img]http://p6.itc.cn/images01/20201120/8b2f6b5ecd564007b3e27ecbda773874.png[/img][/align][url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱柱子根据实际需要,柱子越长越长分离效率越高,但时间长,柱子内径增大,会使柱效下降。3、载气载气的选择一般是根据检测器和分离要求决定的,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]一般使用N2作为载气,[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]时使用He作为载气,对于分离要求高的会使用H2作为载气,但H2作载气的时候用于ECD检测器。4、进样口温度进样口的温度是根据样品的沸点、热稳定性、进样量、柱温等综合考虑,不一定要达到沸点,但要保证样品能随着载气进入色谱柱而不析出来,进样量小的时候可以降低进样口温度,一般高于柱温10~50℃。5、升温程序升温程序类似于[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]的洗脱程序,起始温度和升温速率对分离的影响比较大,起始温度低有助于提高分离,升温速率低也是,但过低峰形变宽,所以需要综合考虑。相对[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]洗脱程序要容易得多。但终止温度需要保证样品都能够随着载气流出,以免影响下一针的干扰。6、检测器定量常用的检测器是FID,TCD也是通用型的检测器,但灵敏度相对较低。FPD和NPD适合含硫和含氮溶剂的检测,含卤素的溶剂,如氯仿等适合用ECD检测。7、标准溶液和基体植物油的选择实际检测中发现,不同厂家生产的6号溶剂标准溶液组分有一定的差别,有些会残留少量干扰溶剂,检测过程与6号溶剂同时挥发溢出,进而影响残留溶剂的检测结果,有些甚至差异较大。因此,不仅要选择正规厂家购买标准溶液,同时也可以对标液进行预检测,此外每批6号溶剂标液都要重新绘制标准曲线。国标中推荐采用精制植物油或超声脱气后的植物油作为基体植物油,其目的是选择不含有残留溶剂的植物油,统一样品与标准曲线的制备条件,同时消除基质效应。实际操作过程中可以选择压榨植物油,也可以对浸出植物油油进行低温烘烤来让残留溶剂挥发。实验前可以取部分进行仪器检验,确保基体植物油中的溶剂残留已挥发干净。8、平衡时间和温度平衡时间和温度会影响气液两相的动态平衡,对结果的准确性影响很大。平衡时间不够,6号溶剂无法完全从[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]逸入液面空间的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]中,但平衡时间过长会导致油脂基质分解,一般以10min为宜。提高平衡温度有利于增加6号溶剂的响应,提高检测灵敏度。但过高的平衡温度会导致取样时溶剂的冷凝,降低进样精度,所以一般选择50 ~ 60℃。虽然气液两相比例对平衡的影响不显著,但考虑到降低系统误差,称样量要尽量一致,一定要准确在5.00g。9、样品提取方式传统的提取方式采用顶空瓶直接称样,加入提取溶剂和标准溶液后密封,在顶空装置中振荡或水浴中加热,供色谱进样分析。但目前有研究发现通过塑料离心管来代替顶空瓶,并增加离心操作,回收率为91.0% ~ 106.5%,高于顶空瓶称样法国。原因是样品溶解更充分,而且上方气压更有利于提高回收率。无论采用哪种容器,都要确保其气密性,确保不漏气。
有些标准中提到试剂是农药残留分析纯级,有些是农残级的,我想知道它们有区别吗?
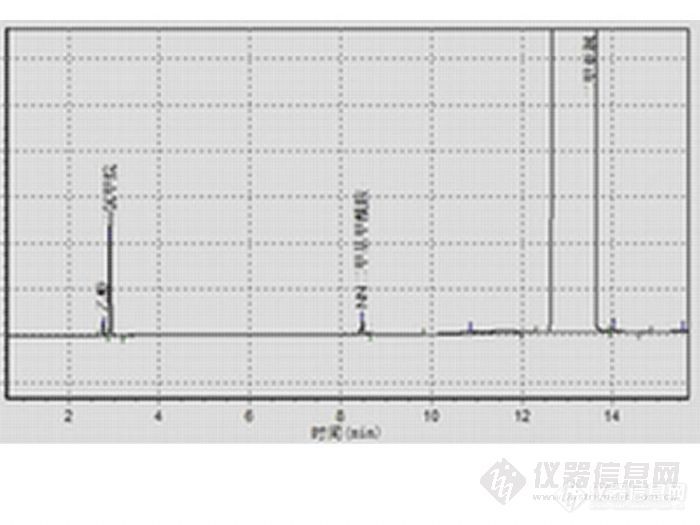
顶空毛细管柱气相色谱法分析测定药品中残留溶剂http://ng1.17img.cn/bbsfiles/images/2011/05/201105211013_295254_2242538_3.jpg 摘要 目前国家对药品中残留溶剂的检测还没有一个统一的国家标准,在关于有机溶剂残留量的指导原则中将二氯甲烷、DMF列为第二类必须控制的毒性试剂,将乙醇、丙酮列为第三类低毒性的溶剂。建立一种快速测定药品中残留溶剂的分析方法很有必要。为此南京科捷采用DK-300A顶空进样器取样,气相色谱毛细管柱分离,分析测定药品中常见的残留溶剂(乙醇、二氯甲烷、NN二甲基甲酰胺、二甲亚枫)。结合相关指标,本实验将二氯甲烷的限量规定为 600ppm,DMF的限量规定为 880ppm,乙醇及丙酮的限量规定为 5000ppm。实验结果表明:顶空毛细管柱气相色谱法快速、简便、准确是测定药品中残留溶剂较为理想的方法。关键词 乙醇 二氯甲烷 NN二甲基甲酰胺 顶空进样 毛细管气相色谱 药品 有机溶剂残留1.药品中乙醇、二氯甲烷、NN二甲基甲酰胺色谱图峰序1. 乙醇2.二氯甲烷3.NN二甲基甲酰胺4.二甲亚枫(溶剂)2.本方法应用范围在合成原料药,辅料或制剂生产的过程中使用或产生的挥发性有机化学物质,它们在实际的生产中未能被完全地清除。本方法可应用在药品中残留溶剂的检测中。近年来,药品中残留有机溶剂的毒性和致癌作用日益引起各方面的重视。药品中残留有机溶剂于1997年被美国FDA列为药品监控项目。我国药品中残留有机溶剂检测也越来越受到有关方面的重视。本文初步研究探索采用顶空毛细管柱气相色谱法分析药品中残留挥发性有机溶剂。顶空气相色谱法只将挥发和半挥发的组份引入柱子,可避免非挥发性的物质对系统的污染,样品前处理简便,分析效率高。结果表明,本方法快速、准确、重现性好。3.仪器及试剂配置色谱仪器配置色谱柱及试剂GC5890(FID检测器)毛细管专用柱30*0.32.*0.5乙醇、二氯甲烷各一瓶顶空进样器:DK-300ANN二甲基甲酰胺1瓶N2000色谱工作站(电脑自备1台)二甲亚枫1瓶氢氮氧一体发生器或钢瓶气各一瓶顶空压盖机1台顶空瓶20ml (带塞) 50只
小麦植株、籽粒和土壤中氯氟吡氧乙酸残留分析方法的建立样品以碱性甲醇混合提取液机械振荡提取后,液液分配净化,采用浓硫酸做为催化剂,甲醇做为衍生化试剂,反应后经石油醚提取,GC-ECD法检测。检测条件的确立:Agilent 6890[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]具ECD检测器 色谱柱:HP-5毛细管柱(30.0m×250um×0.25um) 检测温度:柱温起始温度,70℃,保持1min,以20℃/min至240℃,保持6min 进样口温度250℃,检测器温度300℃ 载气:高纯氮气(99.999%),载气流速为1mL/min 进样方式:不分流方式 进样量为2uL。在此条件下氯氟吡氧乙酸的保留时间为10.5 min左右,仪器对氯氟吡氧乙酸的最小检出量为1.0×10-11 g。提取体系:比较了机械振荡法和超声波振荡法两种提取方式不同提取时间的提取效率,确定了机械振荡30min为氯氟吡氧乙酸优化后的提取方法 比较了乙腈、乙酸乙酯、碱性甲醇等3种提取溶剂对氯氟吡氧乙酸提取效率,确定碱性甲醇为氯氟吡氧乙酸在小麦植株、籽粒、土壤中的提取溶剂。衍生化方法:比较了不同甲醇用量、酯化时间和酯化温度等因素对衍生化结果的影响,结果表明,甲醇用量为2 mL,浓H2SO4 1.5 mL,93~98℃水浴条件下酯化时间10 min,较好。优化后方法的添加回收试验结果表明:在0.01mg/kg~0.80mg/kg的添加浓度范围内,小麦植株中氯氟吡氧乙酸的平均回收率为72.3~86.7%,变异系数为3.02~8.59% 籽粒中氯氟吡氧乙酸的平均回收率为77.7~87.3%,变异系数为2.75~7.61% 土壤中的氯氟吡氧乙酸平均回收率为83.6~95.8%,变异系数为2.87~8.46%。该残留分析方法的准确性、精确性均达到农药残留分析的要求。小麦植株和土壤中氯氟吡氧乙酸残留消解动态2008年在安徽、山东两地的田间残留试验结果表明,氯氟吡氧乙酸的消解动态符合一级反应动力学方程。在合肥试验点,小麦植株上氯氟吡氧乙酸田间消解动态方程为C= 0.1226e-0.1171t,半衰期为5.92d 土壤中氯氟吡氧乙酸田间消解动态方程为C = 0.0861e-0.0828t,半衰期为8.37d。在青岛试验点,小麦植株上氯氟吡氧乙酸田间消解动态方程为C= 0.2149e-0.1368t,半衰期为5.07 d 土壤中氯氟吡氧乙酸田间消解动态方程为C = 0.1478e-0.0893t,半衰期为7.76d。在合肥和青岛两地最终残留试验的小麦籽粒和土壤样品中均未有氯氟吡氧乙酸检出。
在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]、[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]仪器使用过程中,我们经常会看见高浓度质控样品(HQC)后面的0h未知样品中有待测物的色谱峰或者在多个空白样品中发现待测物的色谱峰的现象,造成这种现象的原因,有可能是化合物的残留或者是化合物的污染,这些现象在一定程度上会影响实验结果的准度和精度。那如何在方法开发中,解决残留/污染问题?让我们一起来看看下面的文章。1、残留与污染的定义残留:是指前一个样品残留在分析仪器上的残留物质而引起的测定浓度的变化或者是由被测物在进样系统中吸附而造成的现象。任何形式上的残留都可被视为污染。污染:是指为分析监控样品(如空白基质)及实验对照组,给药前及安慰组样品中出现待测物的峰,也可能由和待测物在保留时间及质谱特性上极其相似的不明来源的其他物质产生的“峰”造成。相对于残留,污染具有更多的随机性和多源性,这使它更难以被诊断和纠正。残留和污染会影响方法的精密度和准确度,因此二者都必须被仔细监视和控制。2、药典9012中残留与污染的要求残留的要求应该在方法建立中考察残留并使之最小。残留可能不影响准确度和精密度。应通过在注射高浓度样品或校正标样后,注射空白样品来估计残留。高浓度样品之后在空白样品中的残留应不超过定量下限的20%,并且不超过内标的5%。如果残留不可避免,应考虑特殊措施,在方法验证时检验并在试验样品分析时应用这些措施,以确保不影响准确度和精密度。这可能包括在高浓度样品后注射空白样品,然后分析下一个试验样品。选择性(污染/干扰的要求)该分析方法应该能够区分目标分析物和内标与基质的内源性组分或样品中其他组分。应该使用至少6个受试者的适宜的空白基质来证明选择性(动物空白基质可以不同批次混合),它们被分别分析并评价干扰。当干扰组分的响应低于分析物定量下限响应的20%,并低于内标响应的5%时,通常即可以接受。应该考察药物代谢物、经样品预处理生成的分解产物以及可能的同服药物引起干扰的程度。在适当情况下,也应该评价代谢物在分析过程中回复转化为母体分析物的可能性。3、残留与污染的区别连续进空白多针,如果干扰峰的响应有逐渐降低的趋势,可判断为系统残留;如果干扰峰响应基本保持不变,则可能是系统污染。残留[img=图片]https://file.jgvogel.cn/134/upload/resources/image/356498.png?x-oss-process=image/resize,w_700,h_700[/img]污染[img=图片]https://file.jgvogel.cn/134/upload/resources/image/356499.png?x-oss-process=image/resize,w_700,h_700[/img]4、残留与污染的控制与消除污染可能来源[list=1][*]采样到储存的样品处理过程中的污染。[*]在实验室的样品制备中出现的污染。[*]非实验物质干扰待测物而造成的污染。[/list]例如:空气中的污染:样品制备过程的溶剂挥发会产生进溅及气溶胶,或者分析实验室旁边刚好是制剂试验室又恰巧在做同一药物。流动相或样品污染:实验中,我们也常用进空气针的方法来判断污染和残留是来自进样板还是来自仪器管路中,通过选择适当的排除法去寻找原因可以有效的加快排查的速度。给药、样品采集的污染:待测药物给了对照组的动物或安慰剂组的人,或者对照组的药剂被待测药物污染或高剂量被当低剂量使用。环境因素也会导致污染,如喂食在不同笼中小鼠时产生失误。同样地,对照组和给药组的动物接触时由于互相舐黏有食物的皮毛而污染。样品存储过程的污染:冻存前不正确放置血浆样品造成交叉污染(例如,由水平而不是垂直放置造成的样品管泄漏)。特别是尿液。仪器或试剂的污染:[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url]吸过高浓度的样品,容器未清洗干净。污染解决的解决方法:加强人员培训,加强人员培训,加强人员培训。。。使分析员理解和识别残留和污染及其对生物分析数据的影响的重要性。残留可能来源[list=1][*]系统中的死体积所产生。[*]由于吸附(耗材,管路)导致的残留。[*]在色谱中不完全的洗脱形成的残留。[/list]残留若无法消除,可以合理的安排样品顺序:如参考血药代谢曲线浓度,将低浓度样品安排在前,高浓度样品靠后的方式,或在高浓度样品后增加空白样品来减少残留残留解决方法,还是需要了解化合物的性质合理配置洗针液,减少[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]管路中的死体积等。具体方法可联系作者哦,欢迎来撩,欢迎来撩,欢迎来撩。。。一般多肽化合物或logP值比较高的小分子化合物在方法开发的时候就需要重点考察残留了。5、残留和污染对实验结果的评估如果残留空白中分析物峰面积大于LLOQ峰面积的20.0%,那么残留可能会影响到分析的结果,需要进行残留评估。具体评估方法如下:[img=图片]https://file.jgvogel.cn/134/upload/resources/image/356500.png?x-oss-process=image/resize,w_700,h_700[/img]当样品的残留影响百分比(ECI%)小于5%时,可以认为该样品不受影响,相反则需要对该样品进行重分析或重进样。综上所述,残留可以既是真实或经典的分析物残留,又可以是由吸附或污染引起的残留。前者在很大程度上取决于进样系统的硬性设计。后者通常需要不断监测。
我做手性农药的残留分析,进基质标的时候的峰面积不一样
农药的代谢物与残留毒性 上世纪60年代以来的研究进一步发现,除了农药本身以外,它的代谢产物也会出现残留毒性问题,一系列的事件发生引起了对这个问题的重视。美国的Rode L.(1969)报道,在美国加州农场工人进入喷施过对硫磷几天后的柑橘园发生了中毒事件,研究发现是对硫磷的代谢产物对氧磷引起的,这说明有时农药母体化合物的毒性还不如其代谢产物。这一发现不仅促使加强了对农药残留降解的研究,还引发了对农药降解过程中代谢产物毒性的研究。其后,又陆续发现了一些农药的代谢产物具有比母体化合物毒性更大的情况。如乙酰甲胺磷是一种急性毒性不大的农药 (对大鼠的口服半致死剂量LD50为605~1100mg/kg),因此将它列为相对低毒的农药类别。但喷施到植物上后,乙酰甲胺磷会在植物体内代谢成甲胺磷(对大鼠的口服LD50为20~30mg/kg),其毒性提高了20~50倍。茶叶生产上曾对这个问题展开过讨论,即乙酰甲胺磷究竟适不适合在茶树上使用。现在证明尽管乙酰甲胺磷毒性不高,但喷后几天,会出现一个甲胺磷残留的高峰,甲胺磷又是一种高水溶性的化合物,因此存在很高的风险性,不宜在茶叶生产中使用。乐果也是一种相对低毒的有机磷农药(对大鼠的口服LD50为500~600mg/kg),但当喷施到植物上1~2天后,会氧化成为氧乐果(对大鼠的口服LD50为30~50mg/kg),其急性毒性提高 10倍以上。相类似的有涕灭威农药及其在代谢过程中形成的砜和亚砜代谢物,三唑酮代谢形成的烃基三唑酮,杀虫脒代谢形成的4-氯邻甲苯胺,这些代谢物的形成都明显提高了农药的急性毒性或慢性毒性。这就使得在喷施农药后,除了要进行农药母体化合物的残留测定外,还要对其主要的代谢产物,特别是毒性有提高的化合物进行残留测定。 农药杂质与残留毒性 除了农药母体化合物和主要代谢物外,有时农药中含有的杂质也会产生毒性问题。1976年联合国卫生组织和美国援助巴基斯坦时用马拉硫磷杀蚊治疟疾,由于马拉硫磷中含有的杂质马拉氧磷和异马拉硫磷使得数百人中毒,8人死亡。这一事件促使了对农药杂质毒性的研究。上世纪80年代以来我国已经停止生产、销售和使用滴滴涕农药,但在茶叶中还可以检测到滴滴涕农药的残留。经研究发现,这种滴滴涕的残留主要来自三氯杀螨醇农药。由于三氯杀螨醇的化学结构和滴滴涕非常相似,只相差一个氯原子、一个氢原子,因此在三氯杀螨醇加工工艺中,一些环境条件的变化会使产品中出现滴滴涕成分。据对我国三氯杀螨醇产品的成分分析发现,产品中滴滴涕的含量为3%~13%,因此在喷施三氯杀螨醇防治螨类时会出现滴滴涕的残留。正因为如此,1999年农业部颁布了在茶叶生产中禁止使用三氯杀螨醇的决定。此外许多有机磷农药中的氧化物,二硫代氨基甲酸酯类农药(代森锌等)中的乙撑硫脲都是这个问题的实例。 农药残留分析的特点和要求 农药残留分析是应用现代分析技术对各种食品和环境中的微量和痕量的农药母体化合物和代谢物进行定性、定量分析和测定。农药残留分析属于难度较大的分析类别。 1. 农药残留分析属于微量至超微量分析范畴。在上世纪60年代,农药残留分析一般为ppm级,即从1g样品中需要测出微克级的农药物质。但随着科学的发展和对残留测定要求的提高,检测的要求也相应地提高到ppb级(μg/kg,即十亿分之一),甚至ppt级(ng/kg,即一万亿分之一),如果以1g样品计算,最小检出量就需要相应达到纳克(ng,10-9g)级或皮克(pg,l0-12g)级。这就对检测工作提出很高的要求,既要求测定的仪器有非常高的灵敏度,同时还要求检测方法有非常高的精确度。 2. 对样品的前处理要求极高。正因为农药残留分析是一种微量至超微量的分析范畴,所以对前处理的要求就非常高。所谓前处理就是要将样品中的目标物(农药)尽可能完全地提取出来。由于样品中必然含有各种成分,这些成分会对检测过程有很大的干扰,因此要尽最大可能将样品中的其他成分通过纯化而去除,而将提取样品中的目标物质尽可能完全保留,回收率的高低直接关系分析结果的准确度。 3. 由于分析样品都为未知成分样,也就是说样品中含有几种农药及其浓度均为未知,因此对样品的分析就显得非常复杂。
杀菌剂多残留分析方法 检测农产品中农药残留量是评价其是否超过MRL值的前提,因此农药残留分析方法得到各国的重视。无论是多残留分析(MRMs)还是单残留分析(SRMs),均基于相似的检测步骤,多残留分析由于可同时检测多种农药残留的存在,因此通常是首选方法。公职分析化学家协会(AOAC)的方法是国际公认的多残留分析方法,可用于多种农药残留的检测。该方法通常用水溶性的溶剂提取,紧接着的净化用不溶于水的溶剂进行分配,再用硅胶或弗罗里硅土净化,净化后的提取物用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、高效液相色谱检测。尽管AOAC方法可以检测多达325种农药及相关化合物,但也存在一些不足之处,如效率低,不适于进行筛选分析;有些试剂有毒且用量大;不能分析一些新农药等。针对上述缺陷,一些新的提取、净化方法得到重视和发展,如固相萃取(solid-phase extraction,SPE)、固相净化(solid-phase cleanup,SPC)、基质固相分散萃取(matrix solid-phase dispersion,MSPD)、超临界流体萃取(supercritical fluid extraction,SFE)等。这些技术的突出特点是简便、样量小型化、萃取的广泛性。检测技术上,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]及高效液相色谱仍然是主要的技术手段。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方面,采用微波诱导等离子(microwave induced plasma,MIP)-原子发射检测器(atomic emission detector,AED)及多级质谱的检测技术取得了快速的发展;离子及离子对色谱以及柱后衍生技术的应用是高效液相色谱检测的研究热点,激光诱导荧光检测器也已开始应用。超临界流体色谱(Supercritical fluid chromatography, SFC)在农药残留检测中的成功应用,实现了样品中农药残留的提取、净化、检测一步完成,是当前联用技术的典型代表。2.1 提取从样品中提取残留农药的效果,很大程度上决定于农药的极性及样品基质的类型。提取过程通常将样品置于匀浆瓶中,添加溶剂,利用匀浆器(homogeniser)、搅拌机(blender)或超声波发生器(sonicator)匀浆。常用的有机溶剂有丙酮、乙腈、甲醇、乙酸乙脂等,根据样品类型、含水量及目标农药,有时需要添加适量的水或调整pH值。多数情况下样品能均匀的分散在有机溶剂中,从而可提高被测残留物的回收率,减少共提的干扰物比率。经均化作用后,以过滤或离心的方法将溶剂和固体分开。2.1.1乙腈提取法乙腈提取法可应用于大多数农药和其它一些化合物的提取,然而在该方法中,许多水溶性(极性)化合物在石油醚从乙腈水中提取农药以及在弗罗里硅土柱层析或氧化镁/硅藻土柱进行层析净化时,水溶性化合物或部分或全部损失。尽管如此这种提取方法仍适用于许多杀菌剂的分析。在Liao等人的方法中采用乙腈进行提取,通过添加氯化钠使乙腈与水分离,上层部分(乙腈)浓缩至小体积后用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]检测。AOAC早期的方法及美国加州食品和农业部 (California Department of Food and Agriculture,CDFA)的MRSM (Multiresidue Screen Method) 方法多采用乙腈作为溶剂。2.1.2丙酮提取法由于丙酮具有无毒、易于纯化、与乙腈和其它一些溶剂相比挥发性好而且价格低廉等优点,因此也是一种常用的提取溶剂,许多方法采用它。此外,在样品中含糖时,丙酮不会象乙腈那样与水形成两相,故可用于高含糖量的样品。实验表明,丙酮具有广泛的化合物和样品基质适用性,已有的回收率数据包括400余种化合物,其中含杀菌剂40余种。理论上,丙酮可用于提取任何样品中除了带有永久离子之外的残留农药。因此,许多国家农药残留标准方法均采用丙酮作为主要溶剂,如德国的DFG S19方法,美国FDA MRMs方法等。在这些方法中,丙酮提取液用氯化钠或硫酸钠饱和之后,分配至二氯甲烷、己烷或石油醚中,从而可得到对不同化合物有利的分配特性和有机相的快速分离。2.1.3乙酸乙酯提取法乙酸乙酯极性相对丙酮、乙腈要弱,因此其对弱极性农药的提取回收率一般较好些,并且其共提物尤其是色素要显著少于丙酮,从而减少了净化时的压力。采用乙酸乙酯作提取溶剂的方法最早由Ross等提出,提取液采用凝胶渗透层析(GPC)净化(SX-3柱),杀菌剂的回收率超过90%。1989年,瑞典国家食品管理局将其列为国家多残留分析方法,替代了原来由Anderson和Ohlin建议的方法。现在该方法已能检测约160种农药、异构体及降解代谢产物。在荷兰的国家方法中,乙酸乙酯也作为主要的提取溶剂。由于省去净化步骤,乙酸乙酯提取方法也被称为在线提取法(on-line extraction methods),其理论基础是Gibbs三角,可用于在线提取的有机溶剂对还有正己烷/丙酮(8∶2)、乙酸乙酯/二甲苯、丙酮或乙腈/二氯甲烷或石油醚等。2.1.4其它提取方法固相萃取是近年发展较快的一种提取、净化方法,作为一种提取技术,主要应用于液体样品中农药的提取,用于农产品及土壤等固体或半固体样品中农药的提取,实质上是一种净化、富集过程。商品化的固相萃取装置很多,主要是固相萃取小柱、固相萃取盘等,其发展主要体现在填料方面,如石墨化炭黑、键合硅胶、弗罗里硅土、活性炭、硅胶等。基质固相分散萃取是类似于固相萃取的一种提取、净化、富集技术,其是将吸附剂或填料与样品一起研磨分散,然后装柱,用有机溶剂淋洗,使农药淋洗出来,淋洗液一般无需进一步净化,可直接进[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]或高效液相色谱检测。MSPD实际上将包括样品匀浆、细胞裂解、完全提取、分馏及纯化过程集于一个简单的过程。有些淋出杂质较多的样品,也可进行净化、分析。Kadenski等采用MSPD技术,研究了多种农药在一些蔬菜、水果中的回收。与传统方法相比,MSPD具有显著的优点,主要表现在,缩短了样品分析周期;减少了样品量,从而极大的降低了溶剂的用量,降低了环境污染的可能性并提高了操作安全性;适用于自动化操作等。
[b]农药残留分析中不同提取溶剂的评价[/b][i]张 艳[/i] 摘要:从提取溶剂的极性和纯化、农药的特性、溶剂选择及样本的性质等方面,对不同农药、不同样本选择不同提取溶剂进行了分析评价。 关键词:农药残留 分析 提取溶剂 评价 农药已广泛应用于农业生产,它在防治病虫草鼠害等方面发挥着显著的作用,保证和促进了农业的发展。但是,如果不科学、合理的使用农药,会造成农产品和环境的污染。随着社会经济的发展和人们生活水平的不断提高,人们越来越重视食品的内在品质、营养成分和安全卫生,农药残留问题已越来越引起人们的关注。农药残留的监督和检测体系是加强农药管理的重要环节,因此建立一个准确、快速、方便可行的检测方法尤为重要。农药残留检测方法的确定主要包括样本的制备、提取、净化、浓缩、检测等方面内容。这里仅对样本提取中不同提取溶剂的选择进行评价。1 提取溶剂的极性提取是将残留在样本中的农药,采用适当的有机溶剂和方法,从样本中分离出来,以供净化后进行测定。提取是农药残留分析步骤中很关键的一步。提取效果的关键是提取溶剂的选择,提取溶剂的选择与待测农药性质、检测方法及样本种类有关。根据“相似相溶”原理,应选择与待测农药极性相似的溶剂,并要求提取溶剂的沸点应为40~50℃,既能溶解待测农药,又不能与待测农药发生反应。同时要考虑检测器检测时的要求。对含水量高的样本,要选择与水能相混溶的溶剂,还应考虑溶剂对样本的渗透能力等,以便将样本组织中的待测农药充分提取出来。在农药残留分析中,根据农药极性、样本性质等选择不同极性的提取剂,常用提取剂按极性由强到弱为水、乙腈、甲醇、乙醇、丙酮、乙酸乙酯、乙醚、二氯甲烷、三氯甲烷、苯、甲苯、环己烷、正己烷、石油醚。在实际应用中最常用的提取剂有乙腈、甲醇、丙酮、二氯甲烷、正己烷、石油醚。以前曾经单独使用一些极性弱的溶剂如正己烷、苯、石油醚等,提取色素和含油脂少的样本,但是提取率低,不能完全提取植物组织中的残留农药,目前已不再单独使用。丙酮作为极性较强并能与水相溶的提取溶剂,能溶解大多数农药,且过滤和溶解都很容易,但丙酮又能大量提取植物组织中的油脂和色素,为下一步净化带来困难,因此,丙酮可作为单一提取溶剂使用,并适用于提取油脂和色素含量少的样本。乙腈作为提取溶剂对油脂和色素提取较少,在有机磷、有机氯、拟除虫菊酯、氨基甲酸酯类等农药的分析中,被AOAC法所采用,也是我国目前常用的提取溶剂。乙腈与丙酮相比,虽然价格贵一些,且浓缩时间长,但可同时提取多种农药残留,且操作简单,是一种首选提取溶剂。甲醇对氨基甲酸酯类农药提取率较高、效果好但对于含淀粉、胶质等较高的某些样本,存在过滤困难的问题。近年来,当单一溶剂不理想时,通常选择两种或两种以上不同极性的溶剂按不同的比例配成混合提取溶剂,以达到较理想的提取效果。表1列出了几种常用有机磷农药在单一溶剂和混合溶剂中的提取回收率,可以看出,丙酮-正己烷(1∶2)、丙酮苯(1∶2)除了对乙拌磷的提取效果较低外,其它均高于单一溶剂丙酮、苯和正己烷。
本人做磷酸苯丙哌林的残留溶剂分析,釆用药典方法。现在是乙醇丶乙醚丶丙酮三个峰很近有些分不开。请帮助我一下吧!
几个关于农药残留分析的资料,送给这个行业的安全卫士![url=http://www.instrument.com.cn/download/shtml/014602.shtml]农药残留速测技术研究进展[/url][url=http://www.instrument.com.cn/download/shtml/014601.shtml]农药残留分析技术进展概述[/url][url=http://www.instrument.com.cn/download/shtml/014600.shtml]农产品农药残留分析技术进展[/url][url=http://www.instrument.com.cn/download/shtml/014599.shtml]农产品农药残留检测技术[/url] [url=http://www.instrument.com.cn/bbs/main.asp?goto_Url=http%3A%2F%2Fwww%2Einstrument%2Ecom%2Ecn%2Fbbs%2Fshtml%2F20051111%2F273463%2Fp]农药污染控制与农残分析技术进展[/url]
农药残留的问题日益受到人们的关注,各种监督抽查的结果不断见诸于报,这不仅体现了我国政府部门对食品安全的重视程度,而且也成为分析行业生存和发展的一个重要机遇!在农药残留的分析检测中,相关的分析检测方法千差万别,比较权威的分析方法有DFG S19、FDA 2905A、EPA SW-846-3640A、EN12393、EN1528、AOAC No.984.21等等。为了适应农药残留检测项目不断增多的需求,满足食品贸易和食品安全的要求,多农药残留分析已经成为一种非常重要的趋势。在实际分析工作中,越来越多的分析工作者发现,在对水果、蔬菜、肉类、粮食、奶类、茶叶、烟草、中药等样品的农药残留分析中,样品的提取物中往往含有大量的高分子量物质,如油脂、糖类、植物腊质、蛋白质、色素等,这些物质会随着进样被带入到色谱进样系统中。由于这些物质沸点较高,因此会在进样口大量聚集,并会随着进样或者分解而缓慢地进入色谱柱,甚至检测器中,造成背景值增高,检测器污染;日积月累会严重影响汽化效率和分离效果。同时这些物质还会吸附目标化合物,导致目标化合物的色谱峰变形,变小,保留时间漂移,甚至不出峰,这些都会给目标化合物的定性和定量工作造成很大的困难。那么样品提取后选用何种方法来净化样品,以减少基质干扰物对目标化合物色谱柱行为的影响,降低噪音水平,减少检测器的污染,提高灵敏度和分析结果的重现性,是每个分析工作者在农药残留分析中所要考虑的一个重要问题。 常见净化方法有吸附色谱、液液萃取、冷冻、酸碱破坏除脂、凝胶色谱等等。其中,液液萃取、冷冻、酸碱破坏除脂等净化方法,多为手工操作,为了保证良好的重现性,对手工操作的要求很高,很多经典的方法均有提及。吸附色谱通常利用不同粒径、不同活性、不同柱径的氧化铝、硅胶、氟罗里硅土、活性炭等柱或几种不同混合柱来净化样品,已有成熟的自动化仪器,但由于受到技术条件的限制,常见的自动化仪器,样品容量有限,现主要应用在进样前的最后一步净化工作上。凝胶渗透色谱(GPC)也被称为空间排阻色谱(SEC)。该方法基于尺寸排阻的分离原理,利用样品中各组分分子大小不同,从而在凝胶中滞留时间不同而达到分离目的。因此,凝胶渗透色谱(GPC)不仅可以用于分离和测定小分子物质,而且还可以用于分析具有相同化学性质但分子大小不同的高分子量物质。针对上述提出的问题,可以预见凝胶渗透色谱(GPC)在多农药残留分析检测中对于样品提取液中高分子量干扰物的去除具有很好的效果。用凝胶渗透色谱(GPC)对样品进行净化分离时,油脂(通常分子量大于600)等大分子物质首先流出,随后是小分子物质(农药,多氯联苯等),而且淋洗溶剂的极性对分离的影响并不起决定作用,特别适合净化含脂和色素的样品。同时,该方法已完全实现自动化,操作过程简单:样品被转移到小瓶中,注入到凝胶渗透色谱(GPC)柱中进行分离。高分子量物质从柱中洗脱出来,被导入废液瓶中;目标化合物被收集在收集盘的样品瓶中以备后续的处理(通常体积较大,50-150ml左右)。随后系统进行自动清洗,为下一个样品做准备。整个过程全密闭控制,如果结合全自动样品浓缩装置,就可以实现最终浓缩定容,可用于直接进样。并且可以配置不同大小的柱子以满足不同样品量的要求,也同样适用于未知目标化合物的净化。凝胶渗透色谱(GPC)作为样品净化的手段之一,在国外已经得到较为广泛的应用,并且已被证明是一种使用最为方便的样品净化技术。该技术可高效地从有机物样品中除去高分子量的干扰化合物如油脂、糖、聚合物、色素和蛋白质等,降低终端分析仪器的维护频率,减少故障的发生,并可以相对延长分析柱的寿命,直接提高工作效率。凝胶渗透色谱(GPC)在我国应用的还不十分普遍,但是随着农药残留分析技术的发展和对农药残留分析检测工作要求的不断提高,可以相信凝胶渗透色谱(GPC)在我国农药残留样品前处理的分离、净化方面将有更广阔的前景。以上介绍的是最新的样品净化技术,有关样品提取、自动定容和转移等新技术限于篇幅没有介绍。如感兴趣者,请登陆www.gzchemart.com
GC-MS型号:赛默飞 Focus DSQ II进样方式:不分流进样一、事件回放:前不久,据有机组技术员反映,实验室的赛默飞GC-MS已经持续一段时间出现残留问题。问题描述概括为:第一针高浓度阳性样品溶液,接着第二针方法空白会有残留(看阳性样品的浓度,一般残留量在检出限以上),第三针方法空白无残留。二、排查过程记录:1.先排查方法空白是否含有“被测物”。排查流程如下:这台赛默飞GC-MS,连续进方法空白5针,并且同一方法空白也用另一台岛津GC-MS验证。结果:岛津GC-MS结果表明,方法空白是ND;赛默飞GC-MS的5针方法空白,结果都差不多,也是ND。结论:排除方法空白含有“被测物”。2.确认阳性样品后一针的残留浓度是否“稳定”。排查流程如下:这台赛默飞GC-MS,采用“工作溶液曲线最高点走一针”+“方法空白走两针”的进样形式,循环走3次。结果:第一针方法空白都有残留,并且残留量稳定(上一针阳性溶液浓度一定时)。第二针方法空白都是ND。结论:①确认阳性样品溶液后一针的残留浓度“稳定”。②经过上面共两个排查程序,可以肯定整个进样系统里有污染。污染来源可能在于:进样前后洗针程序的次数不够(导致无法洗干净进样针)、衬管填的玻璃棉太厚、吹扫流量太低(或吹扫系统有问题,这台赛默飞GC-MS使用已有近9年的时长)、进样针已坏(导致内部难以洗干净或者有间隙等等,备注:一开始认为这个可能性几乎为零,也拆下来手动清洗发现无异样)。已能够排除的污染来源:洗液、洗针小瓶的软垫、装废液小瓶的软垫、进样口处的隔垫、进样口前端的柱子。下面开始进行剩下污染来源因素的排查。3.排查洗针程序、吹扫流量。排查流程如下:将进样前后的洗针程序修改为:丙酮、正己烷两种溶剂进样前后各清洗5次。将原方法的吹扫流量由50ml/min修改为200ml/min。采用“工作溶液曲线最高点走一针”+“方法空白走两针”的进样形式,循环走3次。结果:第一针方法空白都有残留,并且残留量稳定。第二针方法空白都是ND。结论:洗针程序、吹扫流量不是引起第一针方法空白有残留的原因。4.排查衬管排查流程如下:重新更换全新的并且已去掉玻璃棉的衬管,在第3次排查程序后修改的方法基础上,采用“工作溶液曲线最高点走一针”+“方法空白走两针”的进样形式,循环走3次。结果:第一针方法空白都有残留,并且残留量稳定。第二针方法空白都是ND。结论:衬管以及玻璃棉不是引起第一针方法空白有残留的原因。当排查工作进行到这里的时候,其实我是不太愿意相信很有可能是由于进样针的问题引起第一针方法空白有残留的,主要是主观认为这个可能性实在是低,并且上文也有提到,将进样针拆下清洗发现并无异样。但为了验证最后一个因素,于是进行了最后一次排查工作,如下。5.排查进样针排查流程如下:重新更换全新的进样针,在第4次排查程序的基础上,采用“工作溶液曲线最高点走一针”+“方法空白走两针”的进样形式,循环走3次。结果:全部的方法空白都是ND。结论:引起本次赛默飞GC-MS高浓度阳性样品溶液后一针有残留的原因,在于进样针已损坏(导致内部难以洗干净或者有间隙等等)。后来想想,旧进样针用的时间已长,平时检测任务重,技术员又不太注意清洗保养进样针,导致最终出现这样的结果。但还好的是,最终排查出原因,也算是给大伙提了个醒。排查到最后,竟然是这样的一个环节出了问题,果真应了这句:排除一切不可能因素后,剩下的再不可能,也是真实的答案。三、后记:千里之堤,溃于蝼蚁,从事实验室分析的人员更应注重细节。希望大家从这次小事件中,也留意下并做好实验室里的各处细节。最后,第一次发帖,献给了这次神奇的GC-MS被测物残留原因分析及排除的过程。
确定可能的残留溶剂: 残留溶剂的检测首先要明确有可能会有哪些溶剂残留,也就是溶剂有可能在哪个环节带入,从原料、制备工艺、处方各个方面都需要考虑可能的残留溶剂,再根据残留溶剂的类型确定残留溶剂的残留量限值。 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]分析方法注意事项 对于溶剂残留检测通常用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]来进行检测,在某些情况也会用到高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]等,而残留溶剂的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测方法的开发相对简单,适用范围广,实际运用中不同的溶剂需要再调整。
各位大虾好,小第有一疑问盼大家给点意见。我做美国药典中的盐酸米托蒽醌中的残留溶剂时,按照厂家给的资料要检测其中的2-甲氧基乙醇,厂家给的方法为直接进样(水做溶剂)。但它的样品浓度为200mg/ml,按照盐酸米托蒽醌的溶解度看,样品是绝对不能完全溶解的,样品溶解后成米糊状,离心处理也处理不到上清液,进样发现样品出峰非常的难看,且不见残留溶剂峰。我们打算自己开发方法用顶空做,但美国药典467中建议2-甲氧基乙醇用直接进样法,各位大虾你有过内似的经历么,2-甲氧基乙醇用顶空法检测后,美国FDA能批准么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP