[color=#444444]正在做一个物质的溶剂残留,测定目标为乙酸乙酯,样品已知易溶于碱性水溶液,微溶于常用有机溶剂(已经做过甲醇、DMF、DMSO的溶解度试验)。但是问题是乙酸乙酯不溶于水,在配置对照溶液的过程中乙酸乙酯会挥发,重复性惨不忍睹。并且碱用的是氨,挥发时也会带走乙酸乙酯,溶液稳定性相当差。[/color][color=#444444]尝试过向碱溶液加过甲醇等未果,依然难以使溶液稳定。[/color][color=#444444]求教:如何选择溶剂或者改善溶剂来使乙酸乙酯稳定于溶液中?是否可以通过萃取来得到乙酸乙酯?具体如何操作[/color]
[table=100%][tr][td]求哪位大神能不能给我提供一个用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]顶空进样法测定乙酸乙酯残留的具体实施方案,我查了好多资料也没弄明白[img=,absmiddle]http://muchongimg.xmcimg.com/data/emuch_bbs_images/smilies/shocked.gif[/img][/td][/tr][/table]
有谁做过柿叶提取物乙酸乙酯残留量的吗?参数怎么设置
顶空法检测乙酸乙酯的残留量,发现检出限较高,怎样解决?
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]要用顶空测青霉素钾盐中的正丁醇、乙酸乙酯的残留,可样品不溶于DMS和DMSO溶剂,能溶于水,但正丁醇和乙酸乙酯不溶于水,能进样检测吗?
最近 在做 丙酮 石油醚60-90 乙酸乙酯 的残留检测但是由于石油醚组分复杂 在分离时 石油醚的 中的一个小峰 总是与 丙酮峰重合高手有没有好办法解决下 使用的是DB-624柱
以前很少做气相,现在要做溶剂残留,主要是测乙酸乙酯,仪器是岛津2014,顶空进样。有以下几个问题,求助各位,先谢谢啦1、溶剂应该选什么,我们目前是用二甲亚砜做溶剂,是否可以,二甲亚砜的沸点比较高,应该比乙酸乙酯出峰要晚吧2、大家有没有用内标,是不是自动进样就可以不用内标呢?3、用什么色谱柱合适,我用的是HP-5的柱子4、气相做样品时,每做完一针是否需要在初始温度平衡一段时间
顶空法检测乙酸乙酯的残留量,发现检出限较高,如何解决?
最近在做一个原料药的残留溶剂测定,其中有乙酸乙酯,甲醇,DMF,冰乙酸等残留溶剂需要测定。经过摸索后选用HP-innowax柱测定这四种残留溶剂,用水做溶剂配制对照品溶液和供试品溶液,直接进样测定。可是乙酸的峰型时好时坏,不知道是什么原因?大家都用什么方法与柱子测定乙酸,已经试过DB-624与HP-5柱,乙酸在这两根柱子上的峰型都不好,请大家帮帮忙吧,试了好久了,都没解决
高手请看,乙酸乙酯在原料药中的残留量是否可以用GC-MS-MS进行检测?现有顶空的气相色谱条件,乙酸乙酯的分子量为88.11g/mol,是否可以用MRM定量呢?是否存在二级MS检测碎片离子的质量范围应在28以上的限制呢?请不要说用GC定量即可,因为我们想用更灵敏的方法来检测,能达到ppb更好。我们现在用GC顶空的方法,定量限已经可以达到30ppm左右,我们正在想用更灵敏的方法进行检测。
请问分析乙醇,乙酸乙酯残留用什么柱子比较好?要求乙醇,乙酸乙酯出现在同一张图上.另外,我前几天用DB624做的还可以,后来乙醇峰前后出现了杂峰,乙醇本身也分裂,不知是检测器问题,还是柱子问题,还是进样口问题?谢谢
[color=#444444]高手请看,乙酸乙酯在原料药中的残留量是否可以用GC-MS-MS进行检测?[/color][color=#444444]现有顶空的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,乙酸乙酯的分子量为88.11g/mol,是否可以用MRM定量呢?[/color][color=#444444]是否存在二级MS检测碎片离子的质量范围应在28以上的限制呢?[/color][color=#444444]请不要说用GC定量即可,因为我们想用更灵敏的方法来检测,能达到ppb更好。[/color][color=#444444]我们现在用GC顶空的方法,定量限已经可以达到30ppm左右,我们正在想用更灵敏的方法进行检测。[/color]
最近做了一个样品,要求测定样品中的乙酸残留量,试了好多柱子,最后选定了DB-1701的中等极性的柱子,可新的问题有出来,乙酸的响应值很低,大概的检测限有将近1500ppm了,哪位大侠有方法能提高乙酸的检测限吗。谢谢!!!!!![em09501][em09501]谢谢,给位大侠的帮忙,还有我们要求是做痕量检测的,有时有几百ppm,也几十个ppm的,且同时还要做乙醇和乙酸乙酯。原来是用直接进样做的,但样品量太大了,经常将仪器的分流出口堵住,现在希望通过顶空做改善一下。本人试过极性柱WAX的但乙醇和乙酸乙酯就份不开了。
今天做乙酸乙酯残留,对照面积分别是702.2,722.5,738.6,790.4,821.2,rsd为6.5%,第一针和最后一针面积相差120左右,但rsd在10%内,这5个对照能采用吗?
我现在要做一个乙酸乙酯残留的溶剂残留,但是标样已经没有了。我想请教一下,用分析纯的乙酸乙酯做对照对系统会有什么损害吗?我知道,那样的话,数据的精确度会打折扣,但是,这对系统,包括柱子和监测器会有什么损害吗?谢谢!da
我现有一产品需要做有机溶剂残留量,可能的残留有机溶剂主要为二氯甲烷,乙酸乙酯,甲醇,乙醇.请问应该用什么样的色谱条件?
昨天做实验发现 往浓硫酸加入乙酸乙酯时 没有分层而是乙酸乙酯全部溶到浓硫酸里面。这时什么原因呀?浓硫酸能氧化乙酸乙酯吗?请高手指教?谢谢!
我现在用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做溶剂残留检测,柱子是1701,以甲醇为溶剂,检测乙醇和乙酸乙酯,升温条件为30℃(1min)以15℃/min升至80℃(1min).但是总也分不开。我用SIM模式,检测离子选择46和88
乙酸乙酯浓度在0.025mg/ml,可以在水中溶解,在波长210nm下检测不出来
小麦植株、籽粒和土壤中氯氟吡氧乙酸残留分析方法的建立样品以碱性甲醇混合提取液机械振荡提取后,液液分配净化,采用浓硫酸做为催化剂,甲醇做为衍生化试剂,反应后经石油醚提取,GC-ECD法检测。检测条件的确立:Agilent 6890[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]具ECD检测器 色谱柱:HP-5毛细管柱(30.0m×250um×0.25um) 检测温度:柱温起始温度,70℃,保持1min,以20℃/min至240℃,保持6min 进样口温度250℃,检测器温度300℃ 载气:高纯氮气(99.999%),载气流速为1mL/min 进样方式:不分流方式 进样量为2uL。在此条件下氯氟吡氧乙酸的保留时间为10.5 min左右,仪器对氯氟吡氧乙酸的最小检出量为1.0×10-11 g。提取体系:比较了机械振荡法和超声波振荡法两种提取方式不同提取时间的提取效率,确定了机械振荡30min为氯氟吡氧乙酸优化后的提取方法 比较了乙腈、乙酸乙酯、碱性甲醇等3种提取溶剂对氯氟吡氧乙酸提取效率,确定碱性甲醇为氯氟吡氧乙酸在小麦植株、籽粒、土壤中的提取溶剂。衍生化方法:比较了不同甲醇用量、酯化时间和酯化温度等因素对衍生化结果的影响,结果表明,甲醇用量为2 mL,浓H2SO4 1.5 mL,93~98℃水浴条件下酯化时间10 min,较好。优化后方法的添加回收试验结果表明:在0.01mg/kg~0.80mg/kg的添加浓度范围内,小麦植株中氯氟吡氧乙酸的平均回收率为72.3~86.7%,变异系数为3.02~8.59% 籽粒中氯氟吡氧乙酸的平均回收率为77.7~87.3%,变异系数为2.75~7.61% 土壤中的氯氟吡氧乙酸平均回收率为83.6~95.8%,变异系数为2.87~8.46%。该残留分析方法的准确性、精确性均达到农药残留分析的要求。小麦植株和土壤中氯氟吡氧乙酸残留消解动态2008年在安徽、山东两地的田间残留试验结果表明,氯氟吡氧乙酸的消解动态符合一级反应动力学方程。在合肥试验点,小麦植株上氯氟吡氧乙酸田间消解动态方程为C= 0.1226e-0.1171t,半衰期为5.92d 土壤中氯氟吡氧乙酸田间消解动态方程为C = 0.0861e-0.0828t,半衰期为8.37d。在青岛试验点,小麦植株上氯氟吡氧乙酸田间消解动态方程为C= 0.2149e-0.1368t,半衰期为5.07 d 土壤中氯氟吡氧乙酸田间消解动态方程为C = 0.1478e-0.0893t,半衰期为7.76d。在合肥和青岛两地最终残留试验的小麦籽粒和土壤样品中均未有氯氟吡氧乙酸检出。
那位兄弟又没有欧盟农残检测的方法(乙酸乙酯法) 啊?
目的前段时间公司有个产品需要检测乙酸残留,样品为淡黄色油状液体,不溶于水,溶于有机溶剂。因产品工艺的改变,反应物料中增加了乙酸,也就增加了乙酸残留这项检测。接到任务首先上网查找乙酸检测的相关资料,功夫不负有心人,终于在网上找到一个液相色谱检测乙酸和丙酸的资料。同时电话咨询了一下以前的朋友,还真有位以前的朋友做过乙酸残留的检测,赶紧索要检测方法吧,朋友回答说:应该会有记录,回去找找看。第二天,好消息来了,还真找到当时记录着的方法。但色谱柱的型号忘了,不管了,就这样也可以省好多事了。网上下载的资料与朋友提供的方法有类似之处,也就说明这个方法希望是比较大的,选择现有常用的几款先试试吧。仪器型号:agilent1200色谱柱选择:1、Ultimate,Polar-RP 250mm*4.6mm*5um2、Ultimate,XB C18 250mm*4.6mm*5um检测条件:检测器: UV,210nm进样体积:20ul 流速:1.0ml/min时间A%B% 0 0100 5.0 01005.1 1000 10.0100010.10100流动相的配制:溶液A:甲醇溶液B:0.5%的磷酸二氢钾水溶液,用磷酸或氢氧化钠调节PH=2.8±0.1样品处理:人家用的是用纯水稀释样品,考虑到我们的样品是有机物质,不溶于水中,既然流动相是甲醇,那就用甲醇溶解吧标样的配制
请问:环己烷、甲苯、乙酸乙脂残留测定选用何种[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱

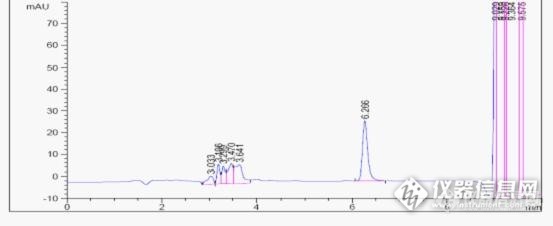
帮我看下乙酸乙酯的标线做的怎么样?1分钟多的是二硫化碳,我用的乙酸乙酯是分析纯的,我推算是1.6分钟那个是乙酸乙酯,其他是杂质峰,老师们觉得呢[img=,690,949]https://ng1.17img.cn/bbsfiles/images/2019/07/201907051351244896_5205_1791505_3.jpg!w690x949.jpg[/img]
我要分析发酵产物中以下几种产物:丙酮、乙醇、丁醇、乙酸和丁酸。用的是“10% Carbowax-20 M, 0.10% H3PO4,support 80/100Chromosorb WAW” 玻璃填充柱和FID检测器。 因为发酵的底物中有一定浓度的糖类物质,所以采取别人的建议在衬管(liner)中填充了石英棉以阻止糖类物质进入柱子,否则糖类物质会很快的污染衬管而出现奇怪的峰。现在发现另外一个问题:那就是乙酸和丁酸总有残留,也就是说在测完一个样后,如果再测一个水样,会出现乙酸和丁酸的峰,而且峰面积比较大。不知道是乙酸和丁酸残留在柱子中,还是liner中或者其他什么地方?是否因为温度不够高?用的方法:进样口温度 225 oC;检测器温度:225 oC;柱温箱(oven)初始温度 40 oC,ramp 1: 40 oC/min for 3min, 最终温度 200 oC for 7 min; 不知有没有人有类似经历,可以给出您的建议解决这个问题?多谢!
[color=#DC143C][size=4]现在乙腈价格飞涨,而乙酸乙酯价格没什么变动,有版友提取用乙酸乙酯代替乙腈进行农残前处理的萃取,这是个不错的建议,不过这个办法可行吗?大家讨论讨论,发表自己的意见。有做过相关实验室的,发表自己的经验有额外的奖励。分享乙酸乙酯提取方法的也将有额外的奖励。[/size][/color]
请问各位工程师,乙酸残留怎么测定?
最近没有事,常想一个问题,我们做金中杂质分析时用乙酸乙酯,而很多单位却用乙醚,大家能告诉我吗?我试着比较两种物质.对比如下1\安全性不用说乙醚危险,极易燃烧,属于管制药品.而乙酸乙酯安全的多,而且满大街都有卖的.2\操作乙醚在萃取过程 ,分层很慢,有的单位做试样时,需要静止5分钟以上,而乙酸乙酯只需要静止半分钟就完全能分离.3\效果乙醚萃取时,如果时间不够长,通常水相颜色很深(萃取不完全),而乙酸乙酯不出现这个问题.4\纯化 乙醚的沸点低,在34度就能进行蒸馏,而乙酸乙酯纯化稍微麻烦,但是在78度蒸馏产物完全能达到萃取效果,而且其中杂质含量和乙醚蒸馏后大致相当.5\金回收由于乙醚沸点较低,按理将其蒸发很容易,然而温度太高,乙醚易着火燃烧,而使金损失严重,另外乙醚直接排出也不利于环保,所以只能通过蒸馏来回收乙醚并分析金.但是在该过程中,蒸馏反应剧烈,可能造成喷溅.相比而言,乙酸乙酯由于沸点高,所以直接在电热板上加热除去乙酸乙酯即可.从上面我觉得应该选择乙酸乙酯,而大多数单位却选择了乙醚,这是为什么呢?
乙酸乙酯沸点77.06℃,折光率1.372 3,相对密度0.9003。乙酸乙酯一般含量为95%~98%, 含有少量水、乙醇和乙酸。可用下法纯化:于1000mL乙酸乙酯中加入100mL乙酸酐,10滴浓硫酸,加热回流4h,除去乙醇和水等杂质,然后进行蒸馏。馏液用20~30g无水碳酸钾振荡,再蒸馏。产物沸点为77℃,纯度可达以上99%。







 我要推广仪器
我要推广仪器
 下载APP
下载APP