哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
尸体在一定条件下,会出现腐败分解反应,这会导致其产生正丙醇、乙醇等物质。在这种情况下,测其血液中的乙醇是不准确的,那么要如何得到乙醇含量的准确值或者其下限?
由于尸体腐败可能会产生乙醇,一般尸体腐败产生乙醇的同时平行产生正丙醇,其正丙醇的浓度是鉴别乙醇是否是死后新生的重要指标。查了下有论文写到这方面,说是研究认为如果血中乙醇含量在正丙醇含量的20倍以内,则可以认定为死后产生的乙醇,否则系生前摄入乙醇。想问下,有谁知道20倍的数据是从哪里来的吗?是有什么标准上面明确写明的吗?
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
尸体在出现腐败分解反应后,会产生正丙醇、乙醇等物质,血样中乙醇受其影响会升高或下降,那么是否会丢失呢?如果是没有饮酒史的人是否会“凭空”产生乙醇?如果会“凭空”产生乙醇,那是溺水死亡的,导致其产生的概率是否会大呢?
GAT 1073-2013 方法测定样品中的乙醇、甲醇、正丙醇、乙醛、丙酮、异丙醇和正丁醇,用的是DB-ALC柱子,请问大家有没这款柱子证书上的TIC图呢?或者自己分析以上目标物的TIC图。。若有,请回复一下,,谢谢
我用的极性柱测甲醇丁醇乙醇异丙醇只出三个峰,50度的程序升温,这是什么原因呢
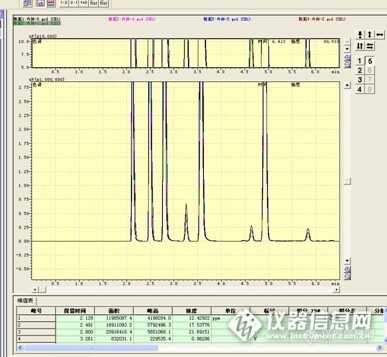
求助一下, ,我这个用程序升温法测甲醇,丙酮,正丙醇(内标) 丁醇 ,丁酯。 110℃ 2.5min 然后25℃/min升到160℃,并保持1min ,,为嘛每次都有三个小峰。如下图片
甲醇有毒,乙醇没毒,丙醇有毒,丁醇有毒,这是我百度的结果,为啥乙醇有毒呢?不明白,是不是除了乙醇没毒,别的都有毒?是不是醇都容易燃烧?
测四氟丙醇中的杂质叔丁醇的含量。因为现有两台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC979/GC1690,手动进样0.8UL,都是FID。柱子都是兰化所的1701毛细柱。分析同一个样品,总峰面积1690为1080W,9790的为998W,结果1690那台叔丁醇的含量是9790的两倍,做了3次试验都是一样情况。两台色谱的载气,空气,氢气的流速都一样。不知道怎样才能确定哪个是准确的。是不是用内标?本人接触色谱不久对内标不是很懂,对叔丁醇选什么内标物好?内标物是不是要特别买?我看试剂的含量都是个范围。
[table=100%][tr][td]使用岛津[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC-2014c TCD检测器, 之前用这个柱子检测过乙醇/水体系和乙二醇/水体系,检测乙醇/水时操作条件为 进样口250° 柱温80°(保留时间30min) TCD检测器250°,检测乙二醇/水体系进样口250° 柱温175°(保留时间30min) 检测器250°。现仍想用此柱子检测正丙醇/水体系,但是不知道柱温怎么设置,正丙醇沸点97.1°,水沸点100°。我测试过如下条件:1.进样口250,柱温60(保留时间30min)检测器250,此条件在1min左右出现两个峰,第一个正丙醇峰未走完接着就出现水峰。并且峰型很不好,倾斜的很严重。2.进样口250,柱温100(保留时间30min)检测器250,此条件只出现一个峰,峰型很好。3.其他 就是柱温 75 °,85°也是连峰,正丙醇峰走完最高点刚往下走了一点点就开始出水峰 。请问各位高手 我该怎么设定 条件?[/td][/tr][/table]
大家好,我想问下正丙醇与四氟丙醇2种溶剂的混合液,怎么检测含量?用气相还是用液相?
同为极性柱子,为什么乙醇叔丁醇在PEG20M的柱子上乙醇出峰在前,FFAP的柱子顺序相反呢?
大家好.我今天查了一下溶剂手册发现上面写着正丙醇的紫外截止吸收波长为210nm,但在我今天在230nm处分析时,发现化学纯的正丙醇有很大的吸收,而且杂峰还比较的多.请问这是怎么回事?[em0715]
我们有发酵液,内含有丙酮、正丁醇、乙醇如何分离,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]参考条件
向各位求教正丙醇的检测方法,不只是纯度,最好能有这方面的国标就更好了!谢谢!
我们样品中要添加正丁醇。所以我们正在分析PTG(聚四亚甲基醚二醇)中正丁醇的含量,一般含量在1200ppm左右。我们分析方法是这样的:用的是内标法,内标物是正丙醇,溶剂是环己烷,内标物的配置是5ml的正丙醇用环己烷稀释到2000 .称取样品2g左右,移取20ml的内表液。摇匀,进样0.2ul.分离也很好,但是重现性很不好。同一个人同一个样品上午和下午作有时能相差200ppm.我现在怀疑环己烷是非极性溶剂,而正丁醇是极性的,环己烷能不能萃取出正丁醇?不知道各位大虾有没有什么好的溶剂溶解PTG萃取出正丁醇?还有内标法对进样量好像要求不是很大,那内标法中有哪些因素影响重复性?很急!!!!!
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]用顶空做样品中的残留溶剂,但是我们先通过检测试过,样品里面没有(也就是检不出)正丙醇的残留,但领导要求还是要做验证,证明你的方法是合适的,请大家帮忙看看应该怎么验证,还有验证的项目有哪些?方案是怎么样的?
为什么在用旋转蒸发仪的时候,有时会加正丙醇来防止爆沸,原理是什么呀?
我们单位买了一台GC-14C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],当时没有调试,我用PEG-20M分离甲醇,乙醇,正丙醇分离度不好,柱效也达不到,请专家指点一下问题在那?
目的:建立一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件同时分离测定环孢素A中乙醇及丙二醇的含量。方法:以GDX-101为固定相,柱长为2 m,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标。结果:乙醇及丙二醇进样量分别在2.0~6.0 μg,1.0~3.0 μg,其峰面积与浓度呈良好的线性关系,加样回收率分别为99.9%(RSD<0.8%,n=5),101.4%(RSD<1.1%,n=5),精密度良好。结论:此[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件可同时测定环孢素A中乙醇及丙二醇的含量,方法简便准确。关键词 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 乙醇 丙二醇 环孢素A山地明(环孢素A)为诺华制药有限公司的产品,是一种免疫抑制剂,用于器官移植和骨髓移植中的抑制排斥现象以及自身免疫疾病。厂方质量标准中乙醇及丙二醇的含量采用石英毛细管柱测定,此种色谱柱在国内使用不普及,我们经多次试验,摸索出一较好的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,适用于国内检测,即以GDX-101为固定相,柱长为2 m,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,氮气为载气,以二甲基亚砜为溶剂,以正丙醇为内标,可同时分离测定环孢素A中乙醇及丙二醇的含量,改进后的方法,乙醇与正丙醇的分离度为3.1,丙二醇与正丙醇的分离度为5.0,符合中国药典1995年版中乙醇量度检查的分离度要求[1],操作简便,结果准确可靠。1 仪器与试药 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:SP-6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱:玻璃柱,长2 m,固定相为GDX-101。 乙醇、异丙醇、丙二醇均为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]纯,二甲基亚砜为色谱纯。 样品:环孢素A胶囊(山地明),由诺华公司提供,批号为187MFD0797;241MFD0797;166MFD0797;483MFD0797;477MFD0797。 标准贮备液及内标贮备液:精密称取[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]级的乙醇及丙二醇2.50及1.25 g分置50 mL容量瓶中,加二甲基亚砜至刻度,摇匀,作为标准贮备液;精密量取正丙醇5.0 mL置50 mL量瓶中,加二甲基亚砜至刻度,摇匀,作为内标贮备液。2 试验方法与结果2.1 色谱条件 采用GDX-101为固定相,柱长为2 m,氮气为载气,采用氢离子火焰检测器,进样口温度为210 ℃,检测器为280 ℃,柱温采用程序升温,即初始为165 ℃,保持12 min,以40 ℃。min-1升至280 ℃,并保持20 min,检测器温度为280 ℃,进样量为2 μL。2.2 分离度试验 称取乙醇、丙二醇及正丙醇各50 mg置同一50 mL量瓶中,加二甲基亚砜至刻度,摇匀,进样2 μL,按上述色谱条件试验,记录色谱图,见图1-A,乙醇、丙二醇及正丙醇的保留时间分别为1.15,2.22,7.54 min,计算乙醇与正丙醇及丙二醇与正丙醇的分离度,其分离度分别为3.1和5.0。图1 分离度色谱(A)及样品测定(B)色谱图1.乙醇 2.正丙醇 3.丙二醇 4.二甲基亚砜2.3 线性范围及标准曲线 分别精密量取乙醇和丙二醇标准贮备液1.0,1.5,2.0,2.5,3.0 mL,分别置50 mL量瓶中,并分别加入内标贮备液1.0 mL,使乙醇终浓度为1.0,1.5,2.0,2.5,3.0 mg.mL-1,丙二醇的终浓度为0.5,0.75,1.0,1.25,1.5 mg.mL-1,分别进样2 μL,以乙醇及丙二醇的进样量为横坐标,以它们的峰面积与内标峰面积之比为纵坐标,分别进行线性回归,结果线性关系良好,乙醇、丙二醇回归方程分别为:A=8.935×103C+7.858×102 r=0.998 8A=8.086×103C-1.649×102 r=0.999 92.4 精密度试验 用乙醇与丙二醇浓度分别2.0及1.0 mg.mL-1的溶液,重复进样5次,结果乙醇与丙二醇的RSD分别为0.7%和1.0%,精密度良好。2.5 回收率试验 采用加样回收法,取已知乙醇与丙二醇含量的样品2粒,用二甲基亚砜溶解,置50 mL量瓶中,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,精密量取此溶液4.0,4.5,5.0,5.5,6.0 mL,分别加入乙醇与丙二醇的浓度分别为2.0 mg.mL-1及1.0 mg.mL-1的标准溶液6.0,5.5,5.0,4.5,4.0 mL,混匀,量取混匀后的溶液2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定这5份溶液的乙醇和丙二醇含量,计算回收率,乙醇的平均回收率为99.9%(RSD<0.8%,n=5),丙二醇的平均回收率为101.4%(RSD<1.1%,n=5)。2.6 样品的测定 取乙醇和丙二醇标准贮备液2.0 mL,内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为对照品溶液;取环孢素A胶囊2粒,置50 mL量瓶中,用二甲基亚砜溶解,精密加入内标贮备液1.0 mL,并加二甲基亚砜至刻度,摇匀,作为样品溶液;分别量取对照品溶液和样品溶液各2 μL,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],按上述色谱条件测定,以内标法计算含量,即得;见图1-B。2.7 对比试验结果 取环孢素A样品5批,用改进后的方法测定样品中乙醇和丙二醇的含量,与厂方测定数据相比,结果基本吻合,见表1。表1 乙醇和丙二醇对比试验结果(%) 批号 本法结果 厂方测定数据 乙醇 丙二醇 乙醇 丙二醇 187MFD0797 101.0 106.3 100.5 105.0 241MFD0797 99.2 99.2 100.6 100.6 166MFD0797 101.7 102.7 101.3 103.0 483MFD0797 98.8 96.8 99.3 97.2 477MFD0797 99.1 98.1 98.9 97.7 3 讨论3.1 本法与原厂方方法相比,方法更为简便,条件普及,有利于对样品质量的控制。3.2 原厂方标准在测定乙醇含量时,以正丁醇为溶剂,由于正丁醇的保留时间与丙二醇过于接近,分离度达不到要求,本法采用二甲基亚砜为溶剂,不影响样品的溶解,同时使丙二醇与二甲基亚砜的分离度符合定量分析的要求。3.3 曾用固定相为GDX-401的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]柱进行检测,乙醇与正丙醇得到完全分离,但丙二醇与溶剂峰重叠,分离度达不到要求。3.4 采用程序升温,可使溶剂出峰时间加快,缩短分析时间。王俊秋(北京市药品检验所 北京 100035)庞青云(北京市药品检验所 北京 100035)余立(北京市药品检验所 北京 100035)参考文献1,中国药典.1995.二部:附录44
帮忙推荐下:能检测醇类(甲醇、丁醇、丙醇等)、氯代烃、丙烯酸、甲基丙烯酸的柱子?
最近在做实验,需要计算手动校正因子。关于有效碳数的计算。想问问大家,异丙醇,甲基叔丁基醚,甲醛,乙醛,丙烯酸,叔丁醇的相对校正因子的计算。望大家给解答下,谢谢。
五种混标用二硫化碳溶解,毛细管柱为安捷伦DB1701,进样口温度250,检测器250,柱温35,程序升温至150,结果只检出甲醇,正丁醇,异戊醇。而乙二醇,异丙醇未检出,是我的仪器条件设置问题吗?求教!
我想用气相色谱分析发酵废液(约85%水分)中丙酮、乙醇、正丁醇含量,用异丁醇做内标液(配的是异丁醇水溶液2.4g/L),5毫升离心后的样品加20毫升内标液进样分析,FID检测器,HP-INNOWAX毛细管色谱柱,30*0.25*0.32,柱温90度,汽化220度、检测器225度,我用分析纯配的标准液峰形很好,但是当进样品时所有峰都拖尾,同一个样品进好多针偶尔有一针峰形还可以。我不知道是不是样品中水分含量太高导致的峰形拖尾,能不能用其他的溶剂配置异丁醇内标液,选择什么溶剂好。谢谢第二个问题:我用上述毛细管色谱柱分析酒精(95%)中杂质含量,杂质有:甲醇、正丙醇、异丙醇、正丁醇(内标物)、异丁醇、异戊醇。程序升温:70度保持3分钟,5度/分钟升到100度,保持2分钟。汽化210度,FID215度。在该条件下除了异丙醇与乙醇分不开所有峰都分离很好,是不是我的色谱柱太短了啊。现在急需要定量异丙醇,有什么好方法,谢谢了
大家好,请问假如我的流动相用的是分析纯的试剂能作液相色谱的流动相吗?比如说我们要使用的流动相是正丙醇和水。我能用分析纯的正丙醇吗?
旋转蒸发时加入正丙醇的作用是什么

目前,许多国家规定,凡结构中具有不对称因素的药物,即“手性药物”,必须拆分其相应的立体异构体,并分别研究其药理、毒理和药物代谢性质。对已上市的消旋体药物,要重新评价其光学异构体的性质。对新申报的药品,一开始就要合成其光学异构体。这就要求我们在产品质量控制过程中,开发相应的光学异构体分离方案。 现大家常用HPLC来进行分离,色谱柱是一部分因素,但是色谱条件的选择,对分离的影响非常大。通常会首先正相条件来进行分离,使用的流动相含有烷烃(正己烷、正戊烷)以及醇类(异丙醇、乙醇、甲醇、叔丁醇),现通过一个案例看叔丁醇在手性分离中的应用。 项目名称是:[b][i]替格瑞洛[/i][/b],结构式分别如下:[img=,416,254]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222022_01_1708019_3.png[/img][img=,374,252]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222023_01_1708019_3.png[/img][img=,388,272]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222023_02_1708019_3.png[/img] 接下来的就是选柱子、选方法了: 当然,开始从正相条件入手,选择了常规的两款多糖涂覆型手性柱:[b]月旭Ultimate Cellu-D(4.6×250mm,5μm)与月旭Ultimate Amy-D(4.6×250mm,5μm)[/b]开始测试。流动相选择了:[b]正己烷:乙醇=90:10[/b],测试效果如下: [img=,690,299]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222030_01_1708019_3.png[/img] [img=,690,301]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222031_01_1708019_3.png[/img] 结果分离效果均不理想,接下来用这两款柱子,醇类选择异丙醇和乙醇按不同比例来测试,均达不到分离要求,但是明显在[b] [/b]Amy-D的分离效果更好,所以后续方法的调整均在Amy-D柱上进行测试; 接下来怎么办呢,试试两种醇混合使用,看看效果: [img=,690,289]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222036_01_1708019_3.png[/img] 上图是使用流动相:[b]正己烷:甲醇:乙醇=90:10:10[/b]来进行分离,结果三个物质均露面了,但是但是还是达不到分离要求,而且降低醇类的比例,也无法实现分离; 哎,看来正相体系是没有办法了,只能再试试模式了。现在[b]极性溶剂模式[/b]也是常用的一种模式,这种模式是使用100%的醇类或两种醇类混合使用,通常用于常规模式分离不理想的时候的一种选择吧!!! 先用[b]甲醇:异丙醇=80:20[/b]开始,出图先: [img=,690,192]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222043_02_1708019_3.png[/img] 分离不开,不行再来,用[b]甲醇:乙醇=90:10:[/b] [img=,690,241]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222045_01_1708019_3.png[/img] 结果三个物质均出现,两个杂质分别在主峰的前后,也达不到基线分离。 继续:100%甲醇、100%乙醇、100%乙腈。。。。。。还是分离不开,咋办。唉这时候已经想选择放弃,通过两个方法,杂质A与杂质B分别控制。 听说有些极端情况下,可以试试叔丁醇呢,那加点叔丁醇试试吧,结果在[b]甲醇:叔丁醇 = 78 : 22[/b]条件下,使用[b]Amy-D(4.6×250mm,5μm)[/b]分析,各个物质均达到基线分离,且杂质A与杂质B均在主峰之前出峰,是不是都是我们希望的,按照要求配置一个系统适应性溶液测试: [img=,690,321]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222048_01_1708019_3.png[/img] 分析条件初步完成,接下来么,就是方法验证了,就不再一一列举了。 所以在进行手性分析时,当遇到分离度不合适,或者样品在烷烃中的溶解性不好以及难以洗脱的样品时,可以试试极性溶剂模式来进行分离,同时因为手性分离,不同醇类,会出现不同的分离效果,而甲醇、叔丁醇也可以当做添加剂的形式,配合其他醇类用于手性分离,有时候会出现意想不到的效果。
安捷伦7890A,顶空自动进样器,19091S-510色谱柱检测乙醇,叔丁醇內标,以前都正常乙醇保留时间为1.2分钟,叔丁醇3.3分钟,现在突然叔丁醇0.6分钟就出峰了,请问一下是什么原因呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP