[em0815] 有些不同的有机溶剂混合后,会形成共沸混合物。哪位能指教一下:什么特征的有机物混合后,可形成共沸混合物?即共沸混合物的形成机理是什么?谢谢!
大家有谁知道双电荷离子的形成过程机理,稠环化合物都有双电荷离子,但是其形成过程和机理不明,有人知道吗?
[font=&]【题名】:电动势形成机理和电极电势的含义[/font][font=&]【全文链接】: https://www.cnki.com.cn/Article/CJFDTOTAL-FXJJ198303001.htm[/font]
求助“内螺纹铜管齿形成形机理的探讨”( 《锻压技术》2005年02期 )
[align=center][b]PBDD/Fs的来源、形成机理、分析方法研究[/b][/align]1 PBDD/Fs的来源PBDD/Fs并不是有意生产的,在各种生产过程中作为不想要的副产品的形式存在。PBDD/Fs来源广泛,包括BFR工艺、电子废物、金属回收和废物焚烧。BFRs的燃烧,特别是PBDE已被报道是PBDD/Fs的一个重要来源。1.1 BFRs产品实际上,40多种溴化有机物可以转化为PBDD/Fs,例如溴酚、TBBPA和PBDEs。在商用多溴二苯醚混合物中,例如BDE-71、BDE-79和BDE-83,PBBs和PBDD/Fs作为杂质以每克数万至数千纳克的浓度存在。PBDD/Fs在商业PBDEs混合物中,浓度大于在PBBs中的浓度。多溴二苯醚和多溴联苯的热解和氧化产生的PBDF/Fs产率很高。分解时,这些BFRs直接转化为PBDD/Fs。几种溴酚类物质的分解,如TBBA、PBDEs和BTBPE,产生了大量的溴苯酚类物质。溴苯酚通过均相和非均相形成PBDD/Fs的有效前驱体。1.2 废旧电器和电子设备电子废物回收场是亚洲发展中国家的大型分布点,它们受到持久性有机污染物的污染,如多溴二苯醚、多氯联苯和PCDD/Fs。传统的处理方法例如加热塑料、电路板和露天燃烧电线等,由回收商进行回收有价值的金属。电子废物中氯化和溴化合物的存在导致在回收过程中产生类似二恶英的化合物。中国的吴荣省环境空气中的PCDD/Fs含量是世界上最高的(64.9-2365 pg-TEQ/m3和0.909-48.9pg-TEQ/m3),同时PBDD/Fs水平也升高,浓度是4-61 pg-TEQ/m3和1.6-2104 pg -TEQ/m3)。除周围空气外,在电子废物拆解区土壤中还发现了2.5-17pg-TEQ/g浓度的PBDD/Fs, 高于或等于全球报告的浓度,浙江省台州市的PBDD/Fs水平也发表了类似的报告。在印度电子废物回收场的土壤中,PBDD/Fs是主要污染物,其浓度在0.27-290 ng/g之间。1.3 冶金工艺工业园区包括烧结厂、EAF、二级ALS和二级铜冶炼厂。在台湾最大的冶金工业园,环境空气中的PCDD/Fs和PBDD/Fs浓度分别为1.7 pg/Nm3(0.15 pg-TEQ/Nm3)和46 fg/Nm3(12 fg TEQ/Nm3)。PBDD/Fs水平高于城市和农村,大气中PCDD/Fs和PBDD/Fs之间的显著相关性表明,冶金工艺也是PBDD/Fs排放的主要来源。冶金工业热工过程的PBDD/Fs水平(0.56-5.8 ng/Nm3)比燃烧过程的PBDD/Fs水平((0.025-0.15 ng/Nm3)高几个数量级。这些结果表明,冶金工艺是PBDD/Fs和前驱物的主要来源。在不同熔炼阶段(进料、熔炼、氧化和脱氧)的PBDD/Fs排放过程中,PBDD/Fs排放主要发生在熔解阶段。1.4焚烧垃圾焚烧是一种减少垃圾、增加土地利用和消除病原体的有效方法,它已成为取代传统垃圾填埋场的主要城市垃圾处理方法之一。在250℃-400℃下,PBDD/Fs的生成速率达到最大值,在325°C,固体中PBDD/Fs的浓度最高。液体和气体样品分别为19000、160000和57 pg TEQ/g,PBDF占主导地位,由于印刷电路板中Br/Cl比值高,PBDD/Fs浓度最高。1.5其他来源根据沉积物样品中PBDD/Fs的时空变化趋势,在工业化前,PBDD比PBDF含量高,这意味着这些沉积物可以自然产生。沿海环境最近的研究还表明,海洋可以产生多个PBDD同系物,海洋环境也是一个重要的天然产生二恶英的环境。人类引起的气候变化和富营养化可能随着藻类和蓝藻的过度生长和活化而导致PBDD/Fs水平的升高。 2 PBDD/Fs的形成途径在过去的二十年中,人们对PCDD/Fs的形成机制进行了研究,包括中式规模实验和适当燃烧条件,氯和溴相似的物理化学性质表明,氯和溴类似的PBDD/Fs和PCDD/Fs具有可比性。几项研究报告说,个人电脑PBDD/Fs可以通过新合成和前驱体化合物形成,如酚类、二苯醚、联苯和多环芳烃(PAHs)。2.1 前体形成相比之下PBDD/Fs的前体生成主要取决于溴化有机化合物(如多溴二苯醚、多溴联苯和TBBP-A),可在热解和焚烧过程中直接形成PBDD/Fs,由PBDE、TBBPA、PBB和PBP形成PBDD/Fs的四个热过程,包括热应力、热解/气化、有效燃烧条件和受控燃烧条件,其中溴化前体产生PBDD/Fs的相关机制是消除反应、缩合反应和脱溴/加氢反应。在焚烧过程中,主要前体之一是PBDE,这需要至少两个步骤来将大多数BFRs转换为PBDD/Fs。对于PBDE,PBDF通常是通过分子内消除邻位Br或邻位H原子然后是环闭合形成的,但是对于PBDD, 分子间的氧插入PBDEs之后是一个环闭合。通常,PBDF的产率比PBDD高出一定的数量级。然而在某些情况下,当存在金属催化剂,如锡、铁、锌、铜或金属氧化物(Sb2O3和Fe2O3)、聚合物基体、水时,PBDD的数量可以超过PBDF。2.2 从头合成与PCDD/Fs类似,在溴存在的情况下观察到了新合成的PBDD/Fs的形成,但这以发现还没有得到广泛的研究。在未来,对PBDD/Fs 的新合成会进行了更详细的研究。生物合成与自然形成一些研究者已经研究了海洋生物的生物合成。文献表明PBDD/Fs可以通过两种主要途径形成,即海洋有机体的生物合成和自然形成。 此外,在海洋环境中会自然产生几类有机溴胺化合物,如溴酚(Bps)、溴氰菊酯、羟基和甲氧基多溴联苯醚等。Haglund等人研究了PBDD/Fs分布的空间趋势,认为PBDDs可以在鱼类和贻贝中自然产生和生物积累。 3 PBDD/Fs分析迄今为止,由于缺乏标准内标和多溴二苯醚的分析干扰,尚未建立一种检测PBDD/Fs的统一分析方法。PBDEs在碰撞诱导解离后能产生一系列碎片离子,如,,和。PBDEs具有相同的m/z值,严重干扰PBDD/Fs的检测。这里介绍了不同的分析方法,包括样品提取和清理、注射技术和色谱分离技术来分析各种基质中的PBDD/Fs。3.1 样品提取有机化合物的主要萃取方法包括索氏提取,加速溶剂萃取(ASE),加压液体萃取(PLE)、液-液萃取、超临界流体萃取和微波辅助萃取,索氏提取和加速溶剂萃取是主要的提取方法。Wyrzykowska等人建议采用软萃取步骤,即先用二氯甲烷然后用甲苯萃取。因为甲苯具有高沸点不适合提取,潜在的热不稳定的溴化分析物,特别是BDE-209。此外,在索氏提取过程中加入铜粉或其他过渡金属催化剂会促进铜催化的脱卤化和多溴联苯醚环化生成PBDD/Fs。一般来说,溴化有机物物比氯化有机物有更多的活性脱卤酶,因此,应保护它们不受光降解(例如紫外光)和热降解的影响。 3.2 净化到目前为止,对于PBDD/Fs的纯化还没有标准的分析程序,大多数研究都提到了对PBDD/Fs进行净化的标准程序。大多数分析人员采用两步或三步色谱柱策略,以避免来自PBDE相同碎片离子的干扰以符合PBDD/Fs测定的要求。一个Florisil柱几乎可以完全分离PBDE和PBDD/Fs,只有不到0.5%的PBDE残留在PBDD/Fs萃取物中。而采用多层硅胶、Florisil硅和活性炭柱相结合的方法测定PBDD/Fs和PBDEs,利用这种技术目标化合物被完全分离,同时只有多层硅胶和Florisil硅柱对基质产生明显的干扰。通过比较硅胶柱和氧化铝柱,有人建议硅胶柱和氧化铝柱能够更有效地从污染的基质中除去一些不需要的物质,因为氧化铝柱对芳香碳具有很高的吸附能,并且对芳烃的分子结构变化很敏感。在日本进行了一项有机溴胺化合物(例如PBDD/Fs和PBDE)的相互校准研究,参与者在一个碳柱上进行了两步清理以实现完全分离。 3.3 PBDD/Fs中的HRGC/HRMS分析目前,正电子电离的高分辨率[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]/高分辨质谱(HRGC/HRMS)由于其高选择性和高灵敏度,被用于PBDD/Fs的分析。另外单离子监测(SIM)是用于增强灵敏度的扫描模式,仪器检测限值(IDL)可达到pg/g。PBDD/Fs的分析受到分析标准、基质干扰、光降解和热降解的限制。EI是HRGC-HRMS的主要的分析方法,少数分析人员使用GC-MS/MS分析PBDD/Fs。GC-MS/MS组合的多重反应监测(MRM)通过直接插入进样针提供专一的选择性和高的灵敏度。与HRGC-HRMS相比,GC-MS/MS的微调谐和维护是它的优势所在。 Wang等人采用GC-MS/MS联用EI模式分析PBDD/Fs。在EI条件下,选择PBDD/Fs中响应强度最高的的分子离子作为母离子,+ 离子用于分析PBDE。线性校准R2≥0.985,方法检出限为0.02-0.4ng/g。为避免分离过程中PBDD/Fs和PBDE的高度降解,通常采用长度为15m或30m的毛细管柱和薄膜厚度(0.1μm或0.25μm)的毛细管柱。. Bjö rklund比较了不同膜厚和柱长对PBDE分析的影响。结果表明采用粗柱时,BDE-209的保留时间增加了2-3 min,而相对面积减少了40%,短柱可以有效地减少热不稳定化合物如的降解如PBDEs和PBDD/Fs。离子源是质谱仪器的核心。Fernando 等人发现EI-MS的方法由于缺乏结构诊断碎片离子而受到限制。尽管CI-MS已被证明是一种高选择性和高灵敏度的分析卤化有机物的技术,但CI不能产生任何含有试剂气体的结构诊断片段。在PBDD/Fs的研究中,Fernando利用负离子大气压化学电喷雾解析PXDD的同分异构体并区分未知的二恶英,但由于缺乏异构体特异性的片段不能区分出来。4 发展趋势目前,发展中国家和发达国家持续使用溴二苯醚的程度尚不清楚,特别是多溴联苯醚,这可能导致人类和环境对PBDD/Fs的接触增加。一个专家小组还建议,应将PBDE和PBDD/Fs作为发展世卫组织技术框架的高度优先事项以及更多的优先事项。PBDD / Fs的时空趋势可以有效地显示出分布、来源和残留水平差异,提供有关其对环境的潜在风险的评价。尽管关于这些趋势的数据仍然非常有限,不管在水生环境中还是在无水环境中,都有必要对这一组化学物质进行进一步的研究。前驱体形成、从头合成、生物合成和自然形成是PBDD/Fs脱溴、缩合或消除的主要途径。由于使用产品多溴二苯醚制剂或其他溴化二苯醚(如TBBP-A和BFR),人为PBDFs大幅增加。因此,为了解释它们的产生过程和生态毒理,不仅需要对人为的而且对自然产生的PBDD/Fs甚至其他类似二恶英的化合物要进行全面的监测调查。

粮食粉尘它属于易燃的粉尘,而从它的理化性质上来分析,粮食还有粮食粉尘很容易就会吸收到热量而不容易散发热量了,粮食还有粮食粉尘局部上也是容易在热传导了,热辐射的作用下或者是本身就发热了,使得了粉尘粒子表面上受热了。[img=,625,917]http://ng1.17img.cn/bbsfiles/images/2017/02/201702041156_01_3190434_3.jpg[/img] 然而这个表面上的额温度上升了,粒子表面上的分子所产生的热分解,就能够形成了环境空气上温度的一种混合产物了,而这样的产物和周围的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]以及固相上的可燃物来继续进行这个化学上的反应了,来放出来热量,那么温度上就很快的增高了,从而使得了反应气体上所发生了强烈的热反应所反映出来的明亮的火焰了,那么就会发生了爆炸。而粉尘爆炸上的形成,通常都是会经过了四个阶段了。易燃易爆粉尘离子在吸收的时候也是要通过外界的热量了,而粒子的表面分子所产生的热分解的阶段了。通常气体上和空气上的乘火车呢过也是混合气体的点火还有局部燃烧的阶段了。
总氮测定加入碱性过硫酸钾之后形成乳白色悬浊液,消解后加入盐酸溶液后乳白色不消失,各位老师知道形成机理吗?怎么能消除?我又取水样加入氢氧化钠试了一下,形成乳白色悬浊液,加入盐酸之后不能变清澈。是什么离子干扰吗?我怀疑是银离子废水中会有那么高的银离子吗?
哪位大侠,可以告诉我稀土元素和哪些有机物形成配合物,其形成配合物的机理是什么?尤其是药物方面,哪些药物可以和稀土元素形成配合物。
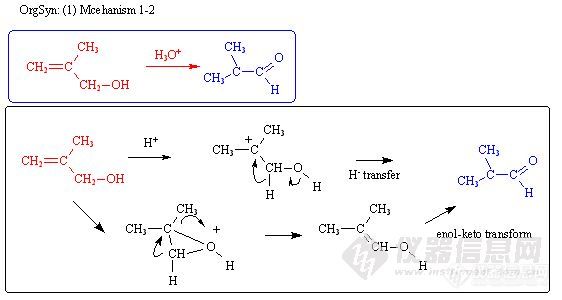
OrgSyn: (1) Mechanism 1-2http://ng1.17img.cn/bbsfiles/images/2011/05/201105151755_294268_1631320_3.jpg基本: 1. 涉及: allylic alcohol的理化性质 2. Driving Force: 酸性中, allyllic alcohol 不稳定 (或易形成 allyllic cation) 3. 思索: hydride transfer 或 enol-keto transform? 是否并存? 上述2机理, 仍没有涉及 allyllic cation. 或许仍有其他路径. 4. 实验探讨: 同位素实验, 使用 –CD2-OH, 看 D 是否完全到产物中? 甚至用同位素氧, 确定是否为原来的 O. 5. 体会: 本反应状似简单, 但存在许多可能因素. 6. 其他: 本题目产物不代表于确定是否为最主要或稳定的产物.进一步说明:本反应机理, 以前多解为 hydride transfer (如上图第一路线), 简单迅速就到达产物.但是也存在另一路径 (我私人观点), 如第二路线所示. 不走 hydride (负氢, H-), 而是让 H 先以质子方式离开, 之后用 enol-keto 转换取回 H.由于反应条件在酸性系统, H- 或容易和 H+ 形成 H-H, 因此偏向于第二路线.由于容易形成 allyllic cation (C=C-C+), 也怀疑产物的 O 是仍为反应物的 O, 或许和溶液中的 H3O+ 有某种程度的交换 (但此交换在机理不必要细述).要确认以上这些, 可以进行同位素试验. 例如将亚甲基 R-CH2-OH 的 H 改成 D, 氧改成氧17 或氧 18.
http://www.hbcnc.edu.cn/~hxx/jpkc/yjhx/dizi.htm点“有机反应机理”,你会看到近百种反应机理。点“有机波谱解析”,直接进行文件下载,下载后是一个可执行程序。有指导性。其中“有机化学实验”也很好,可以参考一下。“有机合成设计”里面没有东西
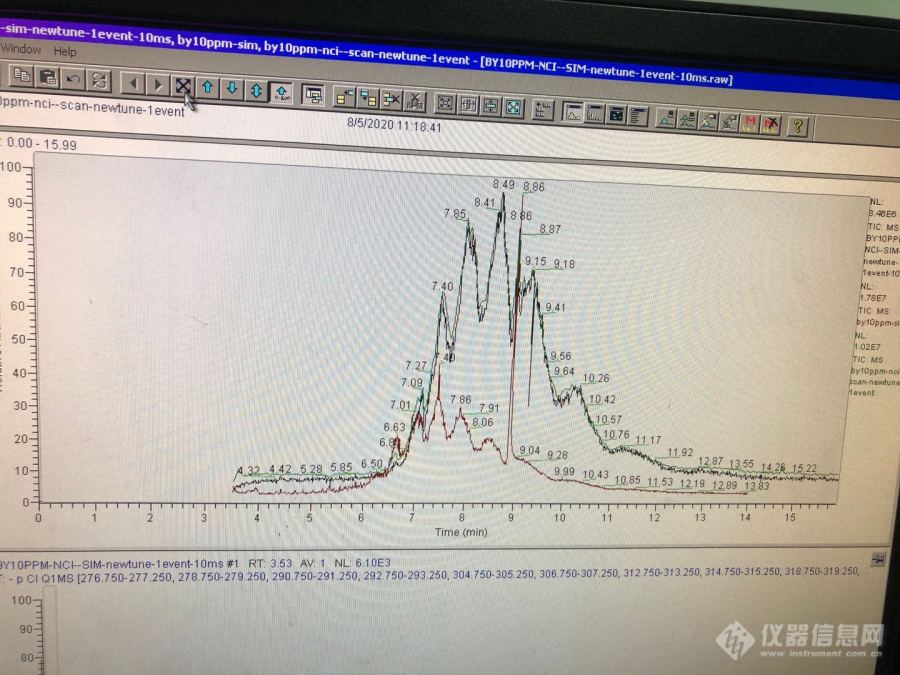
[img=附SCCP的同浓度EI和CI比较图,690,517]https://ng1.17img.cn/bbsfiles/images/2020/08/202008102144382504_8791_2048345_3.jpg!w690x517.jpg[/img]最近尝试用CI源做SCCP,反应气甲烷,一直觉得SCCP在CI下应该是失氯后带正电荷,但事实是形成负离子,还有PCI和NCI分别适合测试哪些物质,归根到底是对CI的反应机理不懂,希望有懂得版友讲解一二或提供资料供学习一下,不胜感激!

[img]http://ng1.17img.cn/bbsfiles/images/2008/11/200811102123_117596_1644912_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2008/11/200811102123_117597_1644912_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2008/11/200811102123_117598_1644912_3.jpg[/img]为什么每片雪花形状都不一样?雪花的形状极多,而且十分美丽.如果把雪花放在放大镜下,可以发现每片雪花都是一幅极其精美的图案,连许多艺术家都赞叹不止。但是,各种各样的雪花形状是怎样形成的呢?雪花大都是六角形的,这是因为雪花属于六方晶系。云中雪花"胚胎"的小冰晶,主要有两种形状。一种呈六棱体状,长而细,叫柱晶,但有时它的两端是尖的,样子象一根针,叫针晶。别一种则呈六角形的薄片状,就象从六棱铅笔上切下来的薄片那样,叫片晶。 如果周围的空气过饱和的程度比较低,冰晶便增长得很慢,并且各边都在均匀地增长。它增大下降时,仍然保持着原来的样子,分别被叫做柱状、针状和片状的雪晶。 如果周围的空气呈高度过饱和状态,那么冰晶在增长过程中不仅体积会增大,而且形状也会变化。最常见的是由片状变为星状。 原来,在冰晶增长的同时,冰晶附近的水汽会被消耗。所以,越靠近冰晶的地方,水汽越稀薄,过饱和程度越低。在紧靠冰晶表面的地方,因为多余的水汽都已凝华在冰晶上了,所以刚刚达到饱和。这样,靠近冰晶处的水汽密度就要比离它远的地方小。水汽就从冰晶周围向冰晶所在处移动。水汽分子首先遇到冰晶的各个角棱和凸出部分,并在这里凝华而使冰晶增长。于是冰晶的各个角棱和凸出部分将首先迅速地增长,而逐渐成为枝叉状。以后,又因为同样的原因在各个枝叉和角棱处长出新的小枝叉来。与此同时,在各个角棱和枝叉之间的凹陷处。空气已经不再是饱和的了。有时,在这里甚至有升华过程,以致水汽被输送到其他地方去。这样就使得角棱和枝叉更为突出,而慢慢地形成了我们熟悉的星状雪花。 上面说的实际上是一个典型的星状雪花的形成过程。它的相当部位,不论形状或大小,都应当是相同的。这种典型的星状雪花只有在一个理想的、平静的环境中(譬如在实验室内)才能形成。在大气中,它不能象上面说的那样有步骤地增大,所形成的形状也就不能那样典型。这是因为冰晶逐渐在下降着,而且有时在旋转着,各个枝叉接触水汽的多少有所不同,而那些接触水汽较多的枝又便增长得较多。因此,我们平常所看到的雪花虽大体上一样但又互不相同。 另外,雪花在云内下降的过程中,也会从适宜于形成这种形状的环境降到适宜于形成另一种形状的环境,于是便出观了各种复杂的雪花形状。有的象袖扣,有的象刺猾。即使都是星状雪花,也有三个枝叉的、六个枝叉的,甚至有十二个枝叉、十八个枝又的。 以上所述都是单个雪花的情况。在雪花下降时,各个雪花也很容易互相攀附并合在一起,成为更大的雪片。雪花的并合大多在以下三种情况下出观。(1)当温度低于0℃的时候,雪花在缓慢下降的途中相撞。碰撞产生了压力和热,使相撞部分有些融化而彼此沾附在一起,随后这些融化的水又立即冻结起来。这样,两个雪花就并合到一起了。(2)在温度略高于0℃的时候,雪花上本来已覆有一层水膜,这时如果两个雪花相碰,便借着水的表面张力而沾合在一起。(3)如果雪花的枝叉很复杂,则两个雪花也可以只因简单的攀连而相挂在一起。 雪花从云中下降到地面,路途很长,在条件适合时,可以经多次攀连并合而变得很大。在降大雪的时候,有时有一些鹅毛般的大雪片,就是经过多次并合而成的。 但是,有时雪花互碰时不是互相并合在一起,而是给碰破了,这时便产生一些畸形的雪花。例如,在降雪的时候,有时会见到一些单个的"星枝",就属于这种情况。
大家好,想跟大家讨论个问题:碱融测硅大家知道碱融后硅酸盐用酸中和成的机理吗?常规测试有两种:硝酸加氢氟酸微波消解,最终形成氟硅酸(部分分解四氟化硅挥发掉,使结果损失),带进去ICP(PE8000)分析后会对雾化器(十字交叉)和矩管有腐蚀,而且记忆效应特别大,以至假的硅数值;第二种用碱融法,使其形成硅酸盐,然后加热水溶解后,加入盐酸或者硝酸中和,使其呈酸性,但理论上在酸性条件下会形成硅酸或者硅胶沉淀,但实际过程并没有沉淀 所以整懵了,在这里提出,跟专家们讨论讨论!
纤维的吸湿过程机理 一般认为纤维吸湿时,水分子先吸附至纤维表面,然后水蒸气向纤维内部扩散,与纤维内大分子上的亲水性基团结合,随后水分子进入纤维的缝隙孔洞,形成毛细水
1、降低的机理氮氧化物 NOx是燃烧过程中氮的各种氧化物的总称,是影响空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量的主要有害成份之一。而正是柴油机燃烧过程中形成的主要物质。它与帆液中的血,红蛋白的亲和力比CO还强,通过呼吸道及肺进入血液,使其丧失输氧能力,刺激人眼粘膜,引起结膜炎、角膜炎,甚至严重时还会引起肺炎和肺气肿、生成的三要素:高温(含局部高温);富氧(含局部富氧);氮与氧在高温下滞留的时间长。这子个要素缺一不可。只有三者同时具备时才能生成,所以,要降低的生成,只要控制其中一个要素,就能奏效。内燃机中乳化柴油燃烧,恰恰在相当程度上遏制了上述子个要素。乳化柴油燃烧时,吸收汽缸内零件的热量以使自身气化,同时减少喷入汽缸内的燃料量;同时,甲醇参与燃烧后的微爆、二次雾化过程以及能减少燃烧辐射热等均使气缸内温度下降并均化,这就能使NOx的排放量下降。2、降低烟度的原理发动机的烟度通常是指炭烟和颗粒及其他排气炯色的统称、它们都是阻光物,义都含有或附有许多有毒成份。内燃机中乳化柴油燃烧时,燃烧进程的活化度增加,燃烧速度加快,燃烧的完全度和完善度提高;同时,由于甲醇的微爆作用使得混合气变均匀等原冈,局部高温和炽热点被消除,缸内温度降低,甚至充量系数也有所增加:所以,柴油机使用含甲醇的乳化油后,烟度都有明显的下降。3、降低CO的机理甲醇在高温下离解成氧及氢氧离子,形成活性中心。它们对燃烧反应,尤其对CO的燃烧反应会起促进作用,这就是高温下甲醇蒸汽的催化作用,,这样,在乳化油燃烧的排烟中,CO含量会明显下降。总之,柴油掺甲醇乳化后,作为燃料不仅节约能源,提高燃烧效率,而且减少环境污染,降低烟气中的氮氧化物、硫氧化物及烟尘含量
[color=#333333]1、降低的机理[/color]氮氧化物 NOx是燃烧过程中氮的各种氧化物的总称,是影响空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量的主要有害成份之一。而正是柴油机燃烧过程中形成的主要物质。它与帆液中的血,红蛋白的亲和力比CO还强,通过呼吸道及肺进入血液,使其丧失输氧能力,刺激人眼粘膜,引起结膜炎、角膜炎,甚至严重时还会引起肺炎和肺气肿、生成的三要素:高温(含局部高温);富氧(含局部富氧);氮与氧在高温下滞留的时间长。这子个要素缺一不可。只有三者同时具备时才能生成,所以,要降低的生成,只要控制其中一个要素,就能奏效。内燃机中乳化柴油燃烧,恰恰在相当程度上遏制了上述子个要素。乳化柴油燃烧时,吸收汽缸内零件的热量以使自身气化,同时减少喷入汽缸内的燃料量;同时,甲醇参与燃烧后的微爆、二次雾化过程以及能减少燃烧辐射热等均使气缸内温度下降并均化,这就能使NOx的排放量下降。2、降低烟度的原理发动机的烟度通常是指炭烟和颗粒及其他排气炯色的统称、它们都是阻光物,义都含有或附有许多有毒成份。内燃机中乳化柴油燃烧时,燃烧进程的活化度增加,燃烧速度加快,燃烧的完全度和完善度提高;同时,由于甲醇的微爆作用使得混合气变均匀等原冈,局部高温和炽热点被消除,缸内温度降低,甚至充量系数也有所增加:所以,柴油机使用含甲醇的乳化油后,烟度都有明显的下降。3、降低CO的机理甲醇在高温下离解成氧及氢氧离子,形成活性中心。它们对燃烧反应,尤其对CO的燃烧反应会起促进作用,这就是高温下甲醇蒸汽的催化作用,,这样,在乳化油燃烧的排烟中,CO含量会明显下降。总之,柴油掺[color=#333333]甲醇乳化后,作为燃料不仅节约能源,提高燃烧效率,而且减少环境污染,降低烟气中的氮氧化物、硫氧化物及烟尘含量。[/color]
小时候在书中看到,在雷雨天气过后,大气中有臭氧的味道,空气感觉特别清新。没想到,现在臭氧也会成为主要污染物,成为危害人类健康的杀手。请教空气中过量的臭氧是怎么产生的,有无去除办法?
浅谈金属钝化的机理我们知道,铁、铝在稀HNO3或稀H2SO4中能很快溶解,但在浓HNO3或浓H2SO4中溶解现象几乎完全停止了,碳钢通常很容易生锈,若在钢中加入适量的Ni、Cr,就成为不锈钢了。金属或合金受一些因素影响,化学稳定性明显增强的现象,称为钝化。由某些钝化剂(化学药品)所引起的金属钝化现象,称为化学钝化。如浓HNO3、浓H2SO4、HClO3、K2Cr2O7、KMnO4等氧化剂都可使金属钝化。金属钝化后,其电极电势向正方向移动,使其失去了原有的特性,如钝化了的铁在铜盐中不能将铜置换出。此外,用电化学方法也可使金属钝化,如将Fe置于H2SO4溶液中作为阳极,用外加电流使阳极极化,采用一定仪器使铁电位升高一定程度,Fe就钝化了。由阳极极化引起的金属钝化现象,叫阳极钝化或电化学钝化。金属处于钝化状态能保护金属防止腐蚀,但有时为了保证金属能正常参与反应而溶解,又必须防止钝化,如电镀和化学电源等。金属是如何钝化的呢?其钝化机理是怎样的?首先要清楚,钝化现象是金属相和溶液相所引起的,还是由界面现象所引起的。有人曾研究过机械性刮磨对处在钝化状态的金属的影响。实验表明,测量时不断刮磨金属表面,则金属的电势剧烈向负方向移动,也就是修整金属表面可引起处在钝态金属的活化。即证明钝化现象是一种界面现象。它是在一定条件下,金属与介质相互接触的界面上发生变化的。电化学钝化是阳极极化时,金属的电位发生变化而在电极表面上形成金属氧化物或盐类。这些物质紧密地覆盖在金属表面上成为钝化膜而导致金属钝化,化学钝化则是像浓HNO3等氧化剂直接对金属的作用而在表面形成氧化膜,或加入易钝化的金属如Cr、Ni等而引起的。化学钝化时,加入的氧化剂浓度还不应小于某一临界值,不然不但不会导致钝态,反将引起金属更快的溶解。金属表面的钝化膜是什么结构,是独立相膜还是吸附性膜呢?目前主要有两种学说,即成相膜理论和吸附理论。成相膜理论认为,当金属溶解时,处在钝化条件下,在表面生成紧密的、复盖性良好的固态物质,这种物质形成独立的相,称为钝化膜或称成相膜,此膜将金属表面和溶液机械地隔离开,使金属的溶解速度大大降低,而呈钝态。实验证据是在某些钝化的金属表面上,可看到成相膜的存在,并能测其厚度和组成。如采用某种能够溶解金属而与氧化膜不起作用的试剂,小心地溶解除去膜下的金属,就可分离出能看见的钝化膜,钝化膜是怎样形成的?当金属阳极溶解时,其周围附近的溶液层成分发生了变化。一方面,溶解下来的金属离子因扩散速度不够快(溶解速度快)而有所积累。另一方面,界面层中的氢离子也要向阴极迁移,溶液中的负离子(包括OH-)向阳极迁移。结果,阳极附近有OH-离子和其他负离子富集。随着电解反应的延续,处于紧邻阳极界面的溶液层中,电解质浓度有可能发展到饱和或过饱和状态。于是,溶度积较小的金属氢氧化物或某种盐类就要沉积在金属表面并形成一层不溶性膜,这膜往往很疏松,它还不足以直接导致金属的钝化,而只能阻碍金属的溶解,但电极表面被它覆盖了,溶液和金属的接触面积大为缩小。于是,就要增大电极的电流密度,电极的电位会变得更正。这就有可能引起OH-离子在电极上放电,其产物(如OH)又和电极表面上的金属原子反应而生成钝化膜。分析得知大多数钝化膜由金属氧化物组成(如铁之Fe2O3),但少数也有由氢氧化物、铬酸盐、磷酸盐、硅酸盐及难溶硫酸盐和氯化物等组成。吸附理论认为,金属表面并不需要形成固态产物膜才钝化,而只要表面或部分表面形成一层氧或含氧粒子(如O2-或OH-)的吸附层也就足以引起钝化了。这吸附层虽只有单分子层厚薄,但由于氧在金属表面上的吸附,改变了金属与溶液的界面结构,使电极反应的活化能升高,金属表面反应能力下降而钝化。此理论主要实验依据是测量界面电容和使某些金属钝化所需电量。实验结果表明,不需形成成相膜也可使一些金属钝化。两种钝化理论都能较好地解释部分实验事实,但又都有成功和不足之处。金属钝化膜确具有成相膜结构,但同时也存在着单分子层的吸附性膜。目前尚不清楚在什么条件下形成成相膜,在什么条件下形成吸附膜。两种理论相互结合还缺乏直接的实验证据,因而钝化理论还有待深入地研究。
等离子消毒灭菌作用机理: 1) 低温等离子的产生及水分子的分解: ◆当水箱等离子发生器产生电流作用时,随着水的分解产生低温等离子。 ◆水分子被分解为氢离子和氧离子之后,又以离子群的形式形成无数小气泡。 ◆在这种形势下,氧原子通过结合其他氧原子,从而显示出其稳定性的特征,并通过结合,形成氧分子。 2) 离子形成过程: ◆电流相互作用形成的能量,使氢原子失去阴离子,同时形成氢的阳离子。 ◆氧分子依附在周围阴离子的电子上,从而形成负氧离子。 ◆这种化学性的电解在空气中大概需要两个小时的时间,在冰箱和其他密闭的环境里大概需要48小时,水的含碱性PH值接近7.4。 3) 氢氧基根的形成过程: 在周围水分子(H2O)的作用下,形成氢阳离子(H+)和氧的阴离子(O-),从而形成氢氧基(OH-)。 同时在这种情况下,形成的过氧化氢(H202)表面蒸发,而氢氧基接触到细菌,有毒化学物质和重金属就产生了杀菌消毒的功效。 4) 杀菌消毒及氧化过程: 氢氧基分子非常不稳定,通过汲取细菌细胞壁上的氢阳离子(H+),有氧化成(H2O)的倾向。 氢氧基(OH-)通过汲取细菌细胞壁上的氢阳离子,从而氧化成水(H2O),同时失去氢离子的细菌也因而氧化成无害物质。 同时,氢氧基能使难以分解的有机物迅速氧化,这种发生的氧化反应能同时消除细菌和苔藓。 另外,水中无数小气泡之间所产生的强大电场也能消除水中的细菌和其他有机/无机物质。
[color=#00008B][size=2][size=4][font=楷体_GB2312][B]病机,是指疾病发生、发展、变化及其结局的机理。以阴阳五行、气血津液、藏象、经络、病因和发病等基础理论,探讨和阐述疾病发生、发展、变化和结局的机理及其基本规律,即病机学说。病机的理论,在《黄帝内经》中已奠定了基础,病机之名,首见于《素问至真要大论》的“审查病机,无失气宜”和“谨守病机,各司其属”。病机的理论在《黄帝内经》中已奠定了基础。如《素问至真要大论》的“诸风掉眩,皆属于肝……”等“病机十九条”,是以“五运六气”的“六气”与五脏相应的理论,将临床常见的诸多症状,分别归属于心、肺、脾、肝、肾之疾患,风、寒、湿、热、火之疾患,病变部位是在“上”或“下”等。但必须指出:《内经》之论述病机,内容非常广泛,并不局限于“病机十九条”,它与邪正和阴阳之盛衰,气血和脏腑之虚实,以及某些病证(如疼痛、痿、痹、厥、痈疽等)的病机,均有详尽的论述。 它的制约因素一是患病机体的体质强弱,二是与致病因素的性质有关。病邪作用于人体,正气奋起抗邪,正邪斗争破坏了人体的阴阳相对平衡,导致阴阳失调,脏腑气机升降失常,气血紊乱,从而产生一系列的病理变化。但从总体来说不外以下几个方面:①邪正斗争。邪正斗争在证候上的反映 ,主要表现虚实的变化 。《素问通评虚实论》说:“邪气盛则实,精气夺则虚。”实,以邪气亢盛为主要矛盾的一种病理反映。一般多见于疾病的早期或中期。如痰涎涌盛,食积不化,瘀血内阻,水湿泛滥及壮热、狂躁,声高气粗,腹痛拒按,二便不通,脉实有力等,均为实证。虚,以正气虚损为主要矛盾的一种病理反映。如大病久病后的神疲体倦,面容憔悴,心悸气短,自汗盗汗或五心烦热,畏寒肢冷,脉虚无力等,均为虚证。邪正斗争还可以概括疾病转归,正胜邪衰则病退,正虚邪实则病进。②阴阳失调。六淫、七情、饮食劳倦等各种因素作用于人体,也必须通过体内的阴阳失调,才能形成疾病。所以,阴阳失调又是疾病发生 、发展的内在依据 。阴阳失调的病理变化,主要表现为阴阳盛衰、阴阳互损、阴阳格拒、阴阳转化,以及阴阳离决等几个方面。③升降失常。人体气的运动可概括为升降出入 ,是脏腑经络 ,阴阳气血矛盾运动的基本过程。因此升降失常,可导致五脏六腑,表里内外,四肢九窍发生各种病理变化。如肺失宣降的胸闷咳喘,胃失和降的呃逆呕恶等。在升降失常中,尤以脾胃升降失调至为重要,因为脾胃是升降运动的枢纽,影响着整体机能活动,所以治脾胃注意调升降是至关重要的原则。 历代医家对于病机学说均非常重视。汉代张仲景的《伤寒杂病论》在《素问》及《灵枢》的基础上,结合临床实践阐述了热病的虚实、寒热、表里、阴阳的进退变化;在《内经》脏腑、经络虚实的基础上,对不少病证的病机进行了阐述。隋代巢元方的《诸病源候论》对1729种病候的病因、病机、及其临床证候作了阐述,成为我国历史上最早的病因病机学专著。金元时期的刘河间在《素问玄机原病式》中提出“六气皆从火化”和“五志过极,皆为热甚”的观点;李东垣在《内外伤辨惑论》中,论述了“内伤脾胃,百病由生”和“火与元气不两立”的病机;张从正在《儒门事亲》中论述了“邪气”致病的病机;朱丹溪在《格致余论》中阐释了“阳有余而阴不足”和“湿热相火”等病机。 病机学说的具体内容可以概括为以下几个方面: 1.从整体上探讨疾病的发生、发展、变化和结局的基本规律。如邪正盛衰、阴阳失调、气血失常、津液代谢失常等。 2.从脏腑、经络等某一系统研究疾病的发生、发展、变化和结局的基本规律。如脏腑病机、经络病机等。 3.探讨某一类疾病的发生、发展、变化和结局的基本规律,如六经传变病机、卫气营血传变病机和三焦传变病机等。 4.研究某一种病证的发生、发展、变化和结局的基本规律,如感冒的病机、哮喘的病机、痰饮的病机、疟疾的病机等等。 5.研究某一种症状的发生、发展的病机。如疼痛的机理、恶寒发热的机理、失眠的机理等等。 6.研究由于气血津液、脏腑等生理功能失调所引起的综合性病机变化,如内生“五邪”。[/B][/font][/size][/size][/color]
[url=https://baike.baidu.com/item/%E6%8A%97%E6%B0%A7%E5%8C%96%E5%89%82/971282?fromModule=lemma_inlink]抗氧化剂[/url]的作用机理是比较复杂的,存在着多种可能性。⑴有的抗氧化剂是由于本身极易被氧化,首先与氧反应,从而保护了食品,如VE。⑵有的抗氧化剂可以放出氢[url=https://baike.baidu.com/item/%E7%A6%BB%E5%AD%90/0?fromModule=lemma_inlink]离子[/url]将油脂在自动氧化过程中所产生的过氧化物分解破坏,使其不能形成醛或酮的产物,如[url=https://baike.baidu.com/item/%E7%A1%AB%E4%BB%A3%E4%BA%8C%E4%B8%99%E9%85%B8%E4%BA%8C%E6%9C%88%E6%A1%82%E9%85%AF/0?fromModule=lemma_inlink]硫代二丙酸二月桂酯[/url]等。⑶有些抗氧化剂可能与其所产生的过氧化物结合,形成氢过氧化物,使油脂氧化过程中断,从而阻止氧化过程的进行,而本身则形成抗氧化剂[url=https://baike.baidu.com/item/%E8%87%AA%E7%94%B1%E5%9F%BA/970597?fromModule=lemma_inlink]自由基[/url],但抗氧化剂自由基可形成稳定的二聚体,或与过氧化自由基ROO–结合,形成稳定的[url=https://baike.baidu.com/item/%E5%8C%96%E5%90%88%E7%89%A9/0?fromModule=lemma_inlink]化合物[/url]。
螯合分散剂在染整加工中阻垢作用机理螯合分散剂的阻垢机理相当复杂,说法也比较多,但下面的说法一直以来是获得最广泛认同的: (一)晶格畸变作用 垢体一般大多为结晶体,以CaCO3垢为例,它的成长是按照严格顺序,由正带电荷的Ca2+与带负电荷的CO3-相撞才能彼此结合,并按一定方向成长。当螯合分散剂在水中加入时,它当中的成分(如有机膦酸成分)物质会吸附到CaCO3晶体的活性增长点上与Ca2+螯合,抑制了晶格向一定的方向成长,因此使晶体歪曲(畸变),长不大,也就是说晶体被螯合分散剂的有机膦酸表面去活剂的分子所包围而失去活性。同样,这种作用也可阻止其它垢类晶体的沉淀。另外,部分吸附在晶体上的化合物,随着晶体增长而被卷入晶格中,使CaCO3晶格发生位错,在垢层中形成一些空洞,分子与分子之间的相互作用减少,使硬垢变软。 而在聚羧酸类螯合分散剂中,聚羧酸是线性高分子化合物,它除了一端吸附在CaCO3晶粒上以外,其余部分则围绕到晶粒周围,使其无法增长而变圆滑。因此晶粒增长受到干扰而歪曲,晶粒变得细小,形成的垢层松软,极易被水流冲洗掉。 (二)增加成垢化合物的溶解度及增溶作用 能与Ca2+、Fe3+、Mg2+等金属离子形成稳定络合物,从而提高了CaCO3晶粒的析出时的过饱和度,也就是说增加了CaCO3在水中的溶解度。另外,由于有机膦酸吸附在CaCO3晶粒增长点上,使其畸变,即相对于不加药剂的水平来说,形成的晶粒要细小的多。从颗粒分散度对溶解度影响的角度看,晶粒小也就意味着CaCO3溶解度变大,因此提高了CaCO3析出时的过饱和度。 (三)静电斥力作用 螯合分散剂的分子在水中电离成阴离子后,由于物理或化学的作用,有强烈的吸附性,它会吸附到悬浮在水中的一些浆料、果胶质、低聚物、染料缔聚体、尘土等杂质的粒子上,使粒子表面带有相同的负电荷,因而使粒子间相互静电排斥,避免颗粒碰撞积聚成长,颗粒呈分散状态悬浮于水中。性能良好螯合分散剂能使颗粒长久地分散在水中,即使产生沉淀,也能减缓颗粒的沉降速度。的如我们自制螯合分散剂ZF,在含有100mg/l钙硬度水中,投加1mg/l,85℃下,可保持24h无沉积。 (四)分散作用 除静电斥力以外,螯合分散剂(如聚丙烯酸)具有分散悬浮作用,能对低聚物、染料缔合体、胶状物等起到强烈分散作用,使其不凝结,加上吸附了螯合分散剂大分子的垢类颗粒产生了空间位阻,呈分散状态的垢类颗粒更不易碰撞凝结而悬浮水中不沉降,易被水冲走。
液相色谱手性识别机理的研究进展黄君珉 陈慧 王琴孙(南开大学元素有机化学研究所 天津 300071)近20年来人们对于用高效液相色谱分离对映体的兴趣与日俱增,发展高效的手性固定相(简称CSP)成为这一领域最活跃的部分,而与之相应的色谱手性识别机理的研究相对来说比较少。但研究色谱拆分机理又是非常重要的,这有利于获得对手性识别更深入的理解,可以指导研制高效的CSPs及预示手性拆分的可能性,而且对理解手性药物的药理、药物设计、生命化学中的立体化学问题等都具有重要意义[1]。物质对映异构体,仅在分子结构上具有不可重叠性。在对称的环境里,无论是气体、固体、或是溶液、液体状态都表现出完全相同的物理化学性质。不管哪一种色谱,为了使互为对映体的物质转化为化学和物理性质不同的非对映体,多宜提供一个手性源,使欲分离的对映体(样品)和手性源(例如:手性固定相)之间形成一个非对映异构分子络合物[2]。非对映分子复合体属于不同的点群。仅对称性的不同,在色谱上是不能被“识别”的,从热力学过程的角度来说,二者必须有一定的自由能差别。1 手性分离的热力学液相色谱手性固定相法直接拆分对映体,在色谱柱内存在着如下的平衡[3]:经典热力学中自由能变化(DG)与焓(DH)、熵(DS)的关系遵从Gibbs方程:DG = DH - TDS 在液相色谱中,保留参数即容量因子k’与溶质在流动相-固定相的热力学平衡常数K的关系为:k’= fK(f是色谱柱相比)。对映异构体选择性a = k’R / k’S (k’R k’S)。色谱过程的自由能变化可以表示成:DG = -RTlnK = -RTln(k’/f) 因此,不难导出:lnK = (-DH/R) × 1/T + DS/R (1) -DR,SDG0 = RT lna = -DR,S DH 0 + TDR,S DS0 lna = (-DR,SDH0/R) × 1/T + DR,SDS0/R (2) 式(1)、(2)表明lnK~1/T、lna~1/T呈线性关系,如图1[4,5]所示:图1 温度对形成非对映异构分子络合物的热力学平衡常数和对映异构体选择性的影响在倒转温度Tinv时,非对映异构分子络合物的热力学平衡常数KR = KS,对映异构体同时流出,在该温度时无对映异构体选择性,aR,S = 1,理论上是由于:lna = (-DR,SDH/R) × 1/T + DR,SDS/R = 0 即: (-△R,S△H/R) × 1/Tinv = DR,SDS/R -DR,SDH = TinvDR,SDS 在该点的两边,温度对对映异构体选择性系数的影响刚好相反,而且溶质对映体流出顺序相反。该点的右边,即:TTinv,色谱手性识别过程为熵变占优势,随着温度的升高,a增大。在手性识别研究中,对映异构体流出顺序在不同温度下倒转的现象迄今只有少量的报道。由于高效液相色谱的温度变化范围较窄,大多数情况下,Tinv不在该温度范围内,并且TTinv,色谱手性识别过程焓变占优势,a值随着温度的升高而降低[6]。手性色谱的对映体分离是一个复杂的色谱过程,Pirkle[7]曾报道了lnK~1/T非线性的实验结果。因此,在不同温度下得到不同的对映体流出顺序也可能是手性色谱保留和拆分机理的改变造成的。图2 N-(3, 5-二硝基苯甲酰基)-亮氨酸正己酰胺及所用手性固定相的结构Pirkle[2]和Davankov等曾分别研究了手性色谱分离对映异构体选择性a和非对映异构体络合物自由能之差(DDG)之间的关系:DR,S DG = -RT lna,考虑到实际的色谱分离过程,非常小的热力学选择性DDG,如果DDG = 0.024 kJ/mol,就可以得到一定的拆分,a = 1.01。随着DDG 的增加,对映体选择性将表现为相应的指数级增长。Pirkle等用实验印证了这种关系[8],在图2所示的CSP上,用30% 异丙醇/正己烷为流动相,N-(3, 5-二硝基苯甲酰基)-亮氨酸正己酰胺的对映异构体选择性测定值a = 10.5,其中(S)-对映体保留较长,相互作用能之差DDG = -5.93 kJ/mol。同样,对具有两个手性中心如图3所示化合物的(SS), (RR)对映体,可以预料:由于手性中心相隔较远,与CSP作用的自由能之差为2DD Gm ,实验所得的手性选择性a为121,大致为前者的平方值a2。后来,Pirkle等再次通过设计出相应的实验提出了这样的论断[9]:具有两个溶质-CSP相互作用部位产生的对映异构体选择性大致为只有一个作用部位所取得的对映异构体选择性值的平方。图3 (SS),(RR)对映体化合物的结构Boehm等[10]用统计热力学理论研究了化学键合手性固定相上溶质对映异构体(AR、AS)的保留行为和分离模式:k’= exp(-bDA) 其中b = 1/(kT),bDA为溶质由流动相到固定相传质过程的Helmholtz自由能,因此:a = ∑iexp(-bEiR)/∑jexp(-bEjS) 其中EiR和EjS分别为R-体(AR)和S-体(AS)在CSP上第i种和第j种作用能。并认为CSP与A的4种一点作用、36种两点作用、12种三点和四点作用中,只有三点和四点作用存在手性识别能力。如果只有一种优势识别模型,则lna 与1/T呈线性关系。Berthod等[11]用热力学方法研究手性识别中手性碳原子所连四个基团各自对手性识别的贡献,分析了126个化合物中81种与手性碳相连的基团,并设氢取代时DG = 0,化合物上各基团独立与CSP作用,在E = ∑|acal - aobs|最小化条件下解如下方程:D(DGA)=(DGA11-DGA12)+(DGA21-DGA22)+(DGA31-DGA32)+(DGA41-DGA42) 定量给出了各个基团对手性识别的贡献(CSP为S-NEC-CD和R-NEC-CD),并发现SP2杂化的碳与手性中心相连比SP3杂化的碳手性识别能力强。该方法只能预示对映体能否被拆分,而不能预示流出顺序。图4 形状选择性的识别模型图5 分子形状和相互作用力共同参与而形成手性识别的模型2 手性识别模型目前,关于手性识别的一般机理众说纷纭。在手性色谱学这一领域,早在1952年,Dalgliesh[12]采用纸层析研究氨基酸对映体的分离时就提出了色谱直接拆分“三点作用”分离理论。后来,Lochmüler和Dobashi提出“两点作用”模型;Lochmüler和Wainer提出“单点作用”机理,Lochmüler进一步提出某些系统存在“环境手性”而没有专一的作用点。对映体的拆分过程可以是熵控制的,手性识别源于形状选择性的识别模型(图4)。也就是说,在没有结合点(如氢键、色散力、偶极作用、p-p相互作用等)的手性环境里,熵控制下,对映体在色谱过程中是可以被拆分的。事实上,由于熵变化值较小,从而导致a 值不够显著,因此,能够通过增加作用点来提高手性选择性值,识别模型如图5所示,对映体的拆分源于分子形状和相互作用力的共同贡献。近些年来,Pirkle等[2]在深入研究手性固定相以及手性色谱立体识别机理的过程中,发展了Dalgliesh观点,再一次阐述了“三点作用”分离理论:手性识别要求手性固定相和对映异构体之间至少有三个同时存在的作用力,这些作用力中至少有一个依赖于立体化学。也就是说,用其中的另一对映异构体(不作任何构象改变)来替代后,至少有一个作用力不复存在或明显改变其性质。用如图6所示的手性识别模型表达:在手性固定相上有三个作用点A、B、C,与之作用的对映异构体也同样有三个作用点A’、B’、C’。对映体I与CSP形成A-A’、B-B’、C-C’三个作用力,对映体II则不存在C-C’作用力。如果C-C’作用力使形成的非对映分子络合物稳定化,那么,色谱分离过程中对映体I比II滞后;反之,对映体I由于C-C’的排斥作用先流出色谱柱。如果C-C’作用力很小,则对映体I、II不能被色谱拆分。图6 色谱手性识别的“三点作用” 模型图7 相似的相互作用力(A-A’, B-B’)导致色谱手性识别能力的降低或消失1992年,Taylor等[13]对“三点作用”原理评述认为:对映体与CSP的三个作用力中,至少有一个力具有立体选择性即依赖于对映异构体和CSP的立体化学,而另外两个作用力必须是两种不同类型的作用力,如氢键、偶极作用、 p-p作用等,否则如果存在两个相同的作用力,则可能产生不利的作用,使得CSP的手性识别能力降低或消失,例如当A-A’和B-B’作用力相同时,就可能使CSP失去手性分离能力(图7)。事实上,手性色谱分离中有的对映体确实是靠氢键这一种类型的力在CSP上识别的[13,14],这种手性识别可以认为是对映异构体和手性固定相形成非对映异构络合物的分子构象不同,使得其平衡常数K1、K2不同。另一方面,“三点作用”原理要求CSP分子和待分离对映异构体的手性中心附近都要有一定的刚性,柔韧性过强将会使手性识别能力丧失,如图8所示。“三点作用”原理与Ogston [15]为解释酶催化反应的立体专一性而提出的“三点键合”原理不同,二者的区别在于:“三点作用”原理没有要求三点都是吸引力。在许多情况下,手性识别可以靠空间位阻的排斥力和两个吸引力来实现(图9),对映体II由于其大基团的空间位阻,使得其另外的氢键和p -p 作用明显减弱。这种作用已被NMR分子间的核极化效应[16]和分子机理计算[3,17]所证实。表现在色谱过程中,对映体II的保留时间会低于对映体I。分子间作用力的单点性和多点性的特征由Pirkle等给出了明晰的描述[2]:两个凸圆面相互接触时,接触处形成一个理想的点,于是将凸圆形电子轨道的相互作用描述成单点性的
求教钢筋锈蚀相关反应机理以及检测方法机理,电位法测试时,有锈蚀电位为什么下降,相关反应有什么?
最近在论坛里多次看到有人问假鸡蛋检测的问题。而我之前真好做过这方面的研究,就把我的看法与大家分享一下。自2001年开始,国内媒体不断报道消费者购买到“假鸡蛋”的现象。2007年,我参加工作进入现在的单位,也多次遇到销售企业要求鉴定鸡蛋真假的业务委托和消费者咨询。这些鸡蛋在煮熟后,蛋黄弹性明显,甚至能摔地弹起而不破碎;掰开后,呈胶质状。但经蛋白质、脂肪、氨基酸、脂肪酸、胆固醇等指标的检测,与普通鸡蛋并无明显区别。当时一养殖企业的负责人问既然不是假的,为什么会这样?我检索了一些资料,最后从上海市地方志办公室商检志网站上找到了一条记录:http://image.sciencenet.cn/album/201305/19/113149q2k1ngacikyb9e8w.png媒体报道“假鸡蛋”的情况依然很多,甚至出现了一些提供“假鸡蛋”制作方法的培训机构。2009年,南京也多次出现“假鸡蛋”,当时省农委一位领导咨询我单位领导是否可以建立一个真假鸡蛋的鉴别检测方法。那时虽然我已基本认定不存在假鸡蛋,但为说服领导(总不能针对一个不存在的东西建个标准),还是撰写了一份技术分析报告上呈领导,基于鸡蛋的生物学特性和结构论证了制造可以以假乱真的“假鸡蛋”是不可能的,成本会非常昂贵,网上流传的“假鸡蛋”制作方法造不出肉眼无法鉴别的假鸡蛋,纯粹是一种培训骗局。一年后央视焦点访谈节目通过记者暗访调查得出了相同的结论。为验证“橡皮蛋”的形成是否有棉籽粕饲料导致,本研究组进行了蛋鸡的棉籽粕饲喂试验,结果显示饲喂棉籽粕后,新鲜鸡蛋蛋黄弹性无明显变化,经储存后逐渐增加,两周后大幅增加,而未饲喂棉籽粕鸡蛋在储存8周后弹性仍无明显变化。对于我们这些畜牧兽医研究者来说,事情到此应该就可以结束了。但我一直在思考为什么棉籽粕会导致“橡皮蛋”的出现?是棉籽粕中的什么物质引发了这一现象?鸡蛋弹性变化的机理是什么?在反复搜索文献后,也只找到两个不同的观点,一个就是上海商检志中提到的由棉酚导致,但没有给出具体机制,也找不到相应的文献佐证。另一个则说鸡蛋的弹性是由棉籽粕中的环丙烯脂肪酸导致,可能机理为环丙烯脂肪酸抑制了肝脏中脂肪酸去饱和酶的活性,而导致蛋黄中饱和脂肪酸含量增加,蛋黄硬度增高。有多篇文章提到这一观点,其内容应来自于国外某学者的研究,但都没有给出相应的参考文献,我也反复检索了英文资料,始终没能找到。http://image.sciencenet.cn/album/201305/19/113204vu9u3k9yn16qyzc3.jpg 但依据这一观点,无法解释为什么鸡蛋要经过一定时间的储存才会出现弹性。而且我们研究中也发现,“橡皮蛋”的饱和脂肪酸含量和普通鸡蛋并没有太大的区别。在反复思考中,我意识到要弄清这一问题,必须先弄清鸡蛋的弹性来自于什么物质的化学变化?(这物质肯定是鸡蛋本身就有的,含量很低的棉酚和环丙烯脂肪酸只可能是诱发了这一物质发生变化导致弹性,而不可能是其自身导致弹性)。还有就是鸡蛋在煮熟过程中发生了什么变化?吃过煮鸡蛋的人都知道鸡蛋煮熟后呈固态,但这其中的机理并没有多少人知道。我也不知道,而且在一番检索后,也没有找到相应的解释。直到有一天,在家给女儿掰果冻的时候,突然顿悟。果冻中含有胶原蛋白,果冻的弹性就来自于胶原蛋白的凝胶化(这些知识实际上一直在脑子里,就是没想到)。而蛋黄的主要成分也就是水、蛋白质和脂肪,如果蛋黄也会发生凝胶化呢,那不就也产生了弹性。(实际上鸡蛋的凝胶化,在工作也多次遇到过,在兽药残留检测时,在鸡蛋中加入碱性乙酸乙酯,涡旋一下,鸡蛋就会变成透明的果冻状,但一直不知道其原因)。拿“凝胶化”和“蛋黄”这两个关键词去google搜索,果然找到一篇文章,是一篇分析食品加工过程中影响蛋黄液凝胶化因素的综述。由浙江大学的陈功和陈有亮老师1999年发表于《肉类研究》杂志。文章中提到鸡蛋的煮熟过程是一个热凝固过程,即天然单体→变性单体→可溶性凝集物或聚合物→凝胶或凝块的过程。蛋黄发生凝胶的关键是蛋白质分子之间发生凝集,形成蛋黄凝胶的蛋白质凝集物主要由低密度脂蛋白(Low-densitylipoprotein, LDL)组成, 而其它蛋白质并不直接参与蛋黄凝胶。对于品质正常的鸡蛋来说煮熟的原蛋黄质地疏松、呈粉状,而搅拌的蛋黄液加热后可形成凝胶,具有一定的弹性、粘合力和硬度。其原因就在于原蛋黄中大部分物质都存在于蛋黄球中,在热凝固过程中被膜的蛋黄球阻碍了蛋白质形成大的凝集物,而搅拌过程破坏了蛋黄球。文章对可影响蛋黄球形态稳定性从而影响蛋黄的凝胶性状产生的因素进行了综述,如渗透压、pH、LDL分子结构和蛋黄
为一个企业领导,在不同的场合,应根据不同的下属,采用不同的激励方法。只有这样,才能收到最大的效果。 激励是不能铁板一块的,它必须根据不同情况灵活实施,体现一个变字。首先,应该根据需要而变。 假设人有五种不同层级的需求,依次为生理需求、安全需求、所属与相爱需求、尊重需求以及自我实现需求。当较低层级的需求获得相当满足,次一层级的需求便会主宰这个人的行为。 这五种需求,事实上没有哪一种需求可能完全得到满足,但是相当程度的满足之后,这一种需求便不再具有激励作用。激励时必须了解被激励者的真实状况,才能够判断他具有什么需求。如果有适当的中介人选,不妨透过中介与被激励者沟通,然后依据他的需求,给予合理的激励。 激励可根据不同的需求,可以采取自助餐式,让不同的被激励者,选择各人的需求;而激励者也要了解不同的对象,施以不同的激励。 组织中不同阶层的成员,也有不同的需求。一般而言,高阶层比较希望大家尊重他,让他觉得自己的确很高明,所以有不同意见,最好不要当面顶撞他,否则他就会恼羞成怒。但是也不能不告诉他,不然他也会怀疑有人要看他的笑话。必须单独委婉地规劝,使其认为自己在改变。 中阶层要告诉他目标,让他自己去找答案,把细节想出来,他才会舒畅。如果给他问题,同时或很快又给他答案,他就会失望,认为自己的能力受到低估。若是他想不出来,可以给他一些启示,还是要他觉得自己找到答案。 基层要清楚一点告诉他应该怎么做,做到什么程度就会满意,最好有工作规范让他按照规定去完成。成果符合标准要表示赞许,使其更加努力。 时间不同,激励的方式也有差异。平常时期按照一般激励,不必采取非常手段。除非发现原来的方法已经日久无效,必须摆脱老一套做法,这才全面更张,改采新的方式,否则不可想到就变,形成特例。
兄弟姐妹们,有没有做反应机理研究的伙伴,想请教一下,做反应机理一定要用同位素标记吗,还有没有可以验证反应途径的方法。谢谢谢谢!!
有机反应机理[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=155416]有机反应机理[/url]
1.机械抛光是一种古老而又最具实用价值的抛光方法,可分为粗抛、中抛和精抛3类: 粗抛是用硬轮对制品表面进行磨削、磨光或研磨,因此粗抛也称为研磨或磨光。它主要用来除去零件表面的毛刺、划痕、锈痕、氧化皮、砂眼、气泡、焊瘤、焊渣和各种宏观的缺陷,以提高表面平整度和降低表面粗糙度。粗抛后的制品表面只能达到平整到平滑的程度,并不能得到光亮的表面,其表面粗糙度在数微米至数百微米之间。 中抛是用较硬的抛光轮对经过粗抛的表面进一步的加工,除去粗抛时留下的划痕,产生平滑至中等光亮的表面。其表面粗糙度在零点几微米到数微米之间。 精抛是抛光的最后一道工序,它是用涂有抛光膏的软轮对零件表面进行加工的方法。由于它是在已经比较平整的表面上进行的,它可以进一步降低表面粗糙度,已达到微观整平的目的,因而可以获得十分光亮的表面,而且抛光时对基材没有明显的磨耗,其表面粗糙度可达到0.01μm左右,可以真正达到镜面效果。2.机械抛光的机理: 机械抛光时,抛光机上的抛光轮在作高速旋转,操作者将被抛光的制件表面以适当压力按压在抛光轮上,这时因摩擦作用而产生高温,使被抛光表面容易发生变形而形成一层“加工变质层”。在旋转着的摩擦力的作用下,一方面表面的某些凸出部分被削去,同时金属制件表面也会产生塑性变形,凸起部分被压入,或移动一段距离后填入凹陷部位。这种削凸填凹的整平过程,以高速度大规模地反复进行,加上抛光膏地光亮化作用,结果就使原来较粗糙地制件表面,变得平滑而光亮。
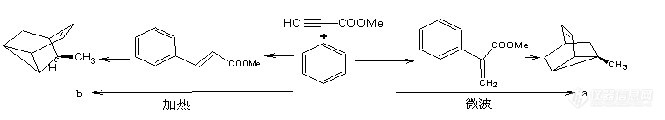
[b]关于微波化学反应机理的探讨[/b][i]苏跃增 孙晓娟 刘萍(江苏石油化工学院化工系 常州 213016)[/i] 微波在化学过程中的功效,愈来愈引起人们的关注;并已将微波用于化学中更多的领域。微波具有比激光低得多的能级,却能在相同的温度甚至更低的温度下,产生比常规方法高几倍甚至几十倍的效率[1],对这种高效率,学术界的观点是不同的,至今尚没有一个严谨的理论能很好地解释微波反应的机理。这无疑制约着微波化学的发展。1 目前对微波影响化学反应机理的认识及局限性 目前,国内外学术界一般认为,微波对化学反应的高效性来自于它对极性物质的热效应:极性分子接受微波辐射能量后,通过分子偶极高速旋转产生内热效应[2],微波对极性分子的热效应是明显的,而传统的加热方式是靠热传导和热对流过程。因而,人们在研究微波反应时,总是将注意力集中在改变微波辐射功率、辐射时间、原料配比、反应容器的大小等方面[2-4]。更重要的一点是,这些研究大都以家用微波炉改装成反应装置,其微波频率是固定不变的(2450MHz),所以也从客观上使人们忽略了微波频率、调制方式等电磁波特性与反应功效是否存在一定的关系,也就是忽略了去研究一定频率的微波对不同极性分子的影响是否相同、不同频率微波对相同极性分子的影响是否一样,忽略了电磁波的相的加载方向不同是否对反应影响不同的研究,如果答案是否定的,那么微波对化学反应的影响就不只是简单的热效应,而还应存在着选择性加热的问题(即物质分子结构与微波频率的匹配关系)、存在着某些特定的非热效应的影响,或者是对分子的活化影响。[img]http://ng1.17img.cn/bbsfiles/images/2006/08/200608211034_24246_1613333_3.jpg[/img]目前的一些实验研究,揭示了一些问题的存在:很多反应在微波条件下副反应增加 有些反应在微波条件下并不比常规加热效果更好 微波可诱导一些选择性反应的发生,如在温和的反应条件下,微波效应能使N-烷氧羰基戊内酰胺选择性优先脱N-烷氧羰基[5],再如Giguere等人[6]对分子间的Diels-Alder反应,进行了研究,在下面反应中: 表现出明显的区域选择性.在通常情况下,简单烯和不对称亲烯体的反应生成异构体混合物,其中烯和亲烯体的b-碳反应所得产物b占优势,但上面的反应式清楚地表明在微波条件下是在亲烯体的a-碳上形成新键,得到产物a,而且未观察到异构体b的生成。 再如,胡希明[7]等人利用微波合成磷酸锌:[img]http://ng1.17img.cn/bbsfiles/images/2006/08/200608211036_24247_1613333_3.jpg[/img]在沸水浴中进行常规反应,不断有氨气放出,产率很低,要提高产率,就必需不断地补充尿素;而在沸水浴条件不变,增加微波辐射的情况下,氨气逸出很少,一次按化学反应计量配比投料,产率即可高达98%。这个现象用过热理论很难解释(如果认为此频率的微波与(NH2)2CO分子结构更为匹配,相当于进行了选择性加热,也降低了反应势能,促使反应;也有人的实验证明:微波有利于(NH2)2CO的分解,促使CO2的溢出,使反应也有利于向正方向进行。这样解释,似乎更为合理);另外,酞菁铜配合物的微波合成和浓硫酸作为璜化剂酞菁铜配合物的微波磺化反应研究,获得了常规加热条件下不能制备的水溶性磺化酞菁铜配合物[8]。这也表现出了微波辐射对化学反应的非热效应。 而银董红等用微波辐射对ZnCl2-HY分子筛催化剂进行了改性研究:用一定量的无水ZnCl2与焙烧制备的HY分子筛充分研磨后,在2450MHz的微波下,辐射下15min,然后将其用于苯甲醚与乙酰氯的酰化反应,发现这种催化剂有良好的初活性[9]。 在微波条件下,天然产物的变旋反应和放射化学反应[10];非溶剂条件下快速合成氨基酸盐[11],如果只用简单热效应解释,也是不圆满的。Alloum A.B. 等人进行干法有机反应[12],将吸附在KSF上的醇和酯混合物,在160W微波照射50min后,产生75%的醛及34%混合酯。而相似条件下,用普通加热方法一点也得不到醛。如此这些用简单的热效应解释,都不能得到满意的答案。 从以上大量的实验现象来看,我们认为,目前对微波化学反应的机理认识还存在着局限性,在微波化学反应中,应该既存在着热效应,还存在着一些有特殊作用的非热效应。







 我要推广仪器
我要推广仪器
 下载APP
下载APP