怎么筛选差异化合物呀?怎么鉴定差异化合物呀?
http://ng1.17img.cn/bbsfiles/images/2012/12/201212111503_411752_2657521_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212111503_411753_2657521_3.jpgGC-MS法测定了挥发油,结果有两个化合物的匹配度较低,我想鉴定,该怎么办。现将两个化合物的图片截图如上,请问该化合物是什么。
利用紫外光谱可以推导有机化合物的分子骨架中是否含有共轭结构体系,如C=C-C=C、C=C-C=O、苯环等。利用紫外光谱鉴定有机化合物远不如利用红外光谱有效,因为很多化合物在紫外没有吸收或只有微弱的吸收,并且紫外光谱一般比较简单,特征性不强。在一些具有大的共轭体系或发色基团的化合物中,紫外光谱可以作为其他鉴定方法的补充。鉴定化合物主要根据光谱图上的一些特征吸收,特别是最大吸收波长[B]λ[/B]max即摩尔吸光系数ε值来进行鉴定。 如果一直化合物在紫外区是透明的,说明化合物分子中不存在共轭体系,不含有醛基、酮基或溴、碘。可能为脂肪族碳氢化合物、胺、腈、醇等不含双键或环状共轭体系的化合物。 如果在210~250nm有强吸收,表示有K吸收带,可能含有两个双键的共轭体系,如共轭二烯或α,β-不饱和酮等。 如果在260~300nm有强吸收(ε=200~1000),则表示有B带吸收,体系中可能有苯环存在。如果苯环上有共轭的生色基团存在时,则ε1000。 如果在250~300nm有弱吸收带(R吸收带),可能含有简单的非共轭并含有n电子的生色团,如羰基等。 如果化合物呈现许多吸收带,甚至延伸至可见光区,可能含有长链共轭体系或多环芳香性生色团。若化合物具有颜色,则分子中含有的共轭生色团或助色团至少有四个(偶氮化合物除外)。 化合物的紫外光谱实际上反映的是分子中发色基团和助色基团的特性,而 不是整个分子的特性,所以,单独从紫外吸收光谱不能完全确定化合物的分子结构,必须与红外光谱、NMR、MS及其他方法相结合,才能得出可靠的结论。但是紫外光谱在推测化合物结构时,也能提供一些重要的信息如发色官能团,结构中的共轭关系,共轭体系中取代基的位置、种类和数目等。
为什么用GCMS鉴定精油里面的化合物,匹配度都好低啊,要怎么确定化合物是什么
通常我们做的化合物都是相关的,我们可以预测化合物的结构,但是我想知道对于新的未知化合物的核磁谱我们如何定标?[em0805]
未知化合物的结构判定是一个复杂工程,四大波谱分析的综合运用也是必不可少的,质谱分析也扮演其中一个重要角色。如果您是这个化合物的合成者,现在有各种质谱可供选择,您认为未知化合物质谱分析的具体过程是怎么样的?怎样快速准确的完成这一分析任务? [color=#DC143C]感谢参与,参与有奖,好的回帖有更多加分。 谢绝灌水!请各位支持![/color]
有机化合物的鉴定方法,谢谢!
合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。
天然有机化合物提取分离与结构鉴定 书已上传到资料中心作者:汪茂田 谢培山 王忠东 等编著 , 43万字,化学工业出版社, 2004年9月出版 本书阐述了天然有机化合物的提取分离方法和结构鉴定技术。全书共分三篇十一章,阐述了如何利用经典及现代提取、分离技术获得所需要的天然有机化合物;结合实例阐述如何用核磁、质谱方法测试鉴定有机化合物的结构,如何完成图谱解析等。内容汇集了编著者长期积累的教学和实践经验,旨在为从事研发天然产物的工作者提供较系统的理论知识和较全面的实用技术。本书可供从事医药、食品、化妆品、生物化工、精细化工和高分子化工等相关行业的科技工作者参考,也可作为高等院校医药、食品、轻工、化工等类专业课程的教学用书。
合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。
想直接用质谱鉴定结构已知的化合物,主要是想看看生成物中有没有该化合物,用液谱检测有3个峰,想问下,直接进质谱能达到上述目的吗?
[font=微软雅黑][size=16px][color=#444444]合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,欢迎关注漫游药化,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。[/color][/size][/font]
天然有机化合物结构鉴定方法摘自:汪茂田等, 化学工业出版社,200420 世纪下半叶,光谱学已成为有机化学的基础课程。30年代发展的紫外(UV)光谱和40年代的红外(IR)光谱为化学家提供了识别有机化合物生色基和官能团的有效方法。研究者可以采用极少量的样品,非破坏性的实验得到有关结构的信息。50年代发展起来的质谱(MS)方法进一步带来革命性的影响,MS实验可给出化合物的分子式,并且通过裂解方式提供分子的结构信息。 对有机结构化学影响最大的谱学方法当推核磁共振(NMR)。它对有机化学的影响是迅速的并且是震撼性的。近50年来,有机波谱学尤其是NMR技术的发展改革了天然产物结构鉴定的方法。波谱技术已成为探究大自然中分子内部秘密的最可靠、最有效的手段。今天波谱学已成为天然有机化学家不可或缺的工具。可以预期,即使在将来的千年岁月它们也一定是必不可少的。随着波谱技术的飞速发展,将会有更多的新技术为化学家所掌握,那时测定天然产物结构将会变得更加容易。 十几年前像HMBC、TOCSY等2DNMR等技术还是波谱学家刚开发的脉冲序列,可是在近几年市售的NMR波谱仪器上这些技术已成为常规方法了,这些技术已成为天然产物结构测定的强有力的工具。众所周知的吗啡(morphine))是1803年由Serturner分离得到的,直到1952年全合成成功才完成了结构确定,用了150年时间,番木鳖碱(Strychnine,士的宁)的结构确定用了半个多世纪(1891-1946),耗费了几代杰出化学家的心血,原因是当时确定结构的主要方法是湿法化学。而今天确定一个比较复杂的天然化合物的结构已变成研究生的科研训练课程,一般只需几个小时、几天、几周或几个月即可完成。这显然得益于波谱技术的发展和普及,同样重要的是一代又一代的天然有机化学家积累的波谱数据可供参考。严格地说,UV和IR属于光谱,MS不是光谱而是物质粒子的质量谱,NMR属于波谱。早年习惯称“四大光谱”,为了方便起见,本编中统称为波谱法。由于NMR技术在天然物结构测定中的重要地位,加之NMR技术解决天然产物结构问题的“多才多艺”,所以本编讨论的重点侧重于NMR方法。 用波谱法鉴定天然化合物结构需要的样品量。在进行天然物化学成分研究时或微量有机合成,一般分离出来的单体都是微量的,其量的范围通常在几mg至几十mg之间。当样品量大于10mg时,测定多种图谱已足够了。建议先测定NMR,因为测定了NMR的样品可以回收。 当只有几个mg样品且样品来之不易时,测定前需要有一个细致的方案。比如样品量约为5mg,取1~2mg 作为留样预防风险,其余3~4mg用于结构鉴定。根据研究者已获得的背景信息,如果该样品可能是已知化合物,测定1H、13CNMR和MS后,将样品回收再测定IR,与文献数据对照。按理,已知化合物鉴定的最方便的方法是找到对照样品和/或其IR图谱,可实际工作中对照品和对照IR图谱并非容易得到,文献中化合物的IR数据往往只报道几个最大吸收,这对鉴定一个化合物是不够的,因为用IR谱鉴定一个化合物要有图谱对照。如果可能是新化合物,尽可能不做燃烧分析而是采用高分辨MS来决定分子式和碎片离子的元素组成,因为测定MS所需样品量极微。完成各种先期必要的NMR图谱测定后,将样品暂时保留在样品管中,以备有疑问时进一步测定NMR,回收样品用来测定IR等。液体样品涂片测定IR后可用溶剂洗脱来回收,固体样品可从KBr 片中把样品回收。 作者曾用7mg二萜化合物进行酸催化重排反应(化学学报 1987,45,871),由甲醇得2.2mg无色结晶,显微熔点仪测定m.p 257~259℃,然后测定EIMS和1HNMR,回收NMR样品管中的样品,测定IR,再回收KBr 片中的样品。将最后的不足2mg样品进行燃烧分析(只能做一组数据)。 当用 2~3mg 样品测定多种NMR图谱时,花费较长的测试时间是不可避免的。建议尽可能使用磁场较高的仪器(如500MHz、600 MHz、800 MHz),因为磁场越高,灵敏度越高,可大大缩短测试时间,同时对化学位移非常相近的峰也能得到满意的分辨,更有利于图谱的解析。 当一个具有特殊意义的样品只有1 mg左右或者更少时,由于样品量甚微会使工作有一定难度。特别是微克级样品的结构测定,其工作本身就有一定难度,除了尽可能使用高磁场NMR谱仪外,使用微量探头(microprobe)或超低温微量探头测定NMR图谱是可取的。当然使用高分辨MS同样是不可少的。如果混合物中有一系列微克级成分需要鉴定,使用LC-NMR和[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]技术应是一个不错的选择。使用LC-NMR技术分离鉴定将在本章第三节的(四)部分介绍。 样品结构的背景信息 在进行结构鉴定之前,尽可能多地获得与样品化学结构有关的各种背景信息是非常必要的,信息量的多寡直接影响结构鉴定工作的速度。从各种植物中分离出来的成分可以说大部分是已知化合物,只要文献充足,这些已知物的结构鉴定一般都是比较快捷和容易的。对于一小部分新化合物,大多是已知骨架上的取代基不同和/或立体化学不同,文献数据对这些新化合物的鉴定起到非常重要的作用。研究中遇到全新骨架新化合物的几率比较低,即使是新骨架的新化合物,其部分结构单元往往也会在其它天然物结构中出现过,学习和积累天然物结构片段的波谱特征和特征数据,对于鉴定新化合物是很有用的,这也需要有文献数据的参考。所以详尽地查阅与所研究内容有关的文献是一件必要的工作。 在没有和/或缺乏背景信息的情况下,可以直接通过多种波谱技术测定化合物的结构(不包括X-ray四圆衍射法)。由于早期的文献受仪器功能的限制,有些数据没有经过可靠方法的证实(尽管结构没问题),波谱数据不一定完全可靠地归属。文献上通常把归属不清的数据用﹡和/或△表示,并注明了数据可以交换,立体化学未确定的基团用折线连接。由于波谱数据完全可靠的归属对于结构的确证和后人的参考有着重要的意义,所以近年来不断有国内外学者利用高场NMR波谱仪对一些复杂的天然产物进行1H和13NMR化学位移完全归属研究的文献发表,这类文章中的波谱数据很有参考价值。 原始文献和“二手文献”。原始文献是指研究者发表的学术论文,实验报告等,是最有参考价值的资料,作者把结构测定使用的方法、实验条件、分析讨论阐述的比较清楚,有的还有图谱。如果对原作者的结论有疑问,在网络发达的今天,可以很方便地和作者取得联系。所谓的“二手文献”指的是综述、专著、和手册等。综述,尤其是国外作者的综述,由于语言文字和信息来源广泛畅通的缘故更具有参考价值。这里并非贬低我国学者的综述文章,因为有一个不争的事实,那就是有些杂志在国内很难找到或根本找不到。质谱的应用
有机化合物结构鉴定与有机波谱学欢迎下载[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=22260]有机化合物结构鉴定与有机波谱学[/url]
[color=#444444]本人用带PFPD检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]鉴定石油中的噻吩类化合物,请问可否用文献中相应化合物(或标准化合物)的保留时间值作为定性鉴定的依据呢?另外,是不是某种化合物的保留时间会因仪器的种类及分析条件不同而不同呢?谢谢[/color]
你们有用TCD鉴定器分析烃类化合物吗?最小检测浓度多少?
有机化合物结构鉴定方法摘自:汪茂田等, 化学工业出版社,2004
ODP嗅闻结果帮助GCMS鉴定痕量化合物ODP嗅闻结果帮助GCMS鉴定痕量化合物_气质联用(GCMS)仪器社区_仪器信息网论坛 (instrument.com.cn)
大家知道哪里有举办化合物结构鉴定的培训?急需这方面的资料。谢谢[em0808]!
刚到手的,和大家分享[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=131927]天然有机化合物提取分离与结构鉴定[/url]
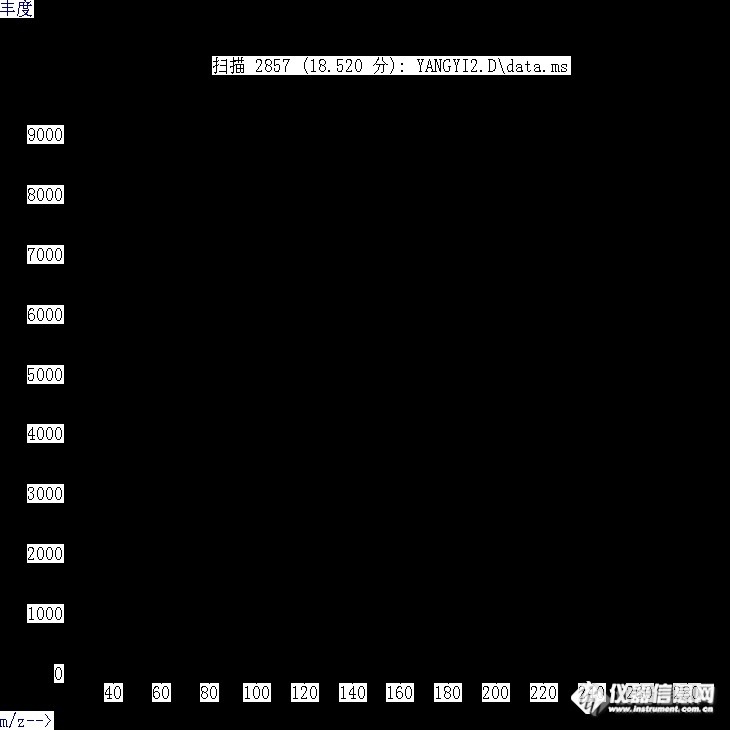
各位请帮忙分析此未知化合物谱图,感谢。[img=,690,261]https://ng1.17img.cn/bbsfiles/images/2019/07/201907030941135926_6185_2680637_3.png[/img]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=25417]有机化合物结构鉴定与有机波谱学[/url]
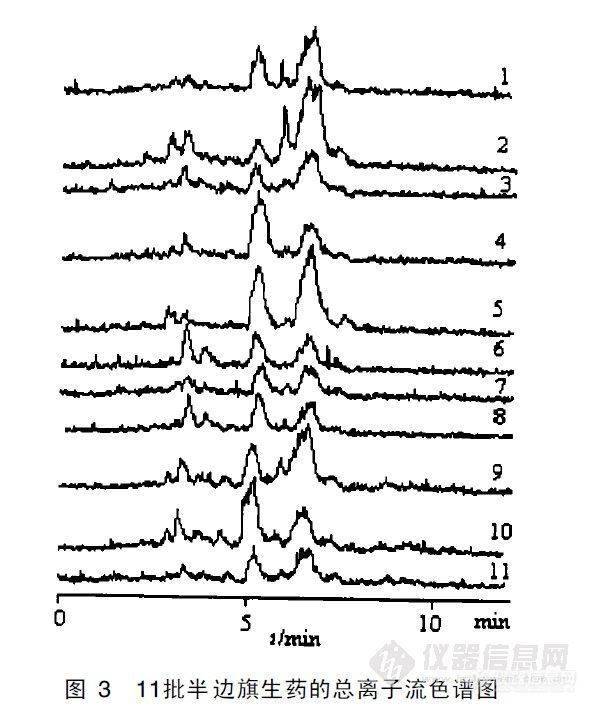
【作者】 邓亦峰; 梁念慈;【Author】 Deng Yifeng,Liang Nianci (Key Laboratory of Natural Drugs Research and Development of Guangdong,Guangdong Medical College,Zhanjiang 524023)【机构】 广东医学院、广东省天然药物研究与开发重点实验室; 广东医学院、广东省天然药物研究与开发重点实验室 广东湛江524023; 广东湛江524023;【摘要】 用高效液相色谱大气压化学电离源质谱联用的方法建立总离子流色谱图,以色谱保留时间和色谱峰的质谱图为依据,对不同地区和不同采集时间的半边旗LC MS图谱中的主要色谱峰进行了对比分析和质谱鉴定。色谱分析条件:色谱柱为DiamonsilODS、4. 6mm×150mm,流动相为30%CH3CN 70% 2mmol/LNH4AC、流速1 0ml/min,进样5μl;质谱分析条件:APCI负离子源,质谱扫描范围m/z50~700,扫描间隔1s。以对照品为参比,鉴定出贝壳杉烷系二萜类化合物4F、5F和6F的色谱峰;根据色谱行为和质谱行为,推测出6F的16位饱和态化合物以及4F和5F的甙。对不同地区和采集时间的半边旗LC MS指纹图谱中的主要色谱峰进行比较分析得出,广州市郊和湛江地区以及在11~12月采集的半边旗中5F含量较高。本法简便、快速,可为半边旗中二萜类抗癌化合物的深入研究和新药资源的开发提供参考。 更多还原【Abstract】 To identify and compare the main peaks of HPLC-APCI-MS FP of the diterpenoids in Pteris semipinnata collected from different region and time, a quadrupole mass spectrometer coupled with atmospheric pressure chemical ionization interface was employed as a detector for HPLC to establish total ion chromatography.HPLC retention time and MS spectrum were used to identify comprehensively.4F,5F and 6F were identified from the chromatography comparing with their standards.The saturated state of 6F and g... 更多还原【关键词】 高效液相色谱质谱联用; 半边旗; 二萜类化合物; 鉴定; 【Key words】 HPLC-APCI-MS; Pteris semipinnata; Diterpenoids; Identify; http://ng1.17img.cn/bbsfiles/images/2012/07/201207302005_380687_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207302005_380688_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207302005_380689_2352694_3.jpg

直接见附件,很强的155和159峰,M+应为358,芳烃馏分。http://ng1.17img.cn/bbsfiles/images/2012/01/201201161521_345955_1615838_3.jpg帮忙鉴定是什么化合物?
有机化合物结构鉴定与有机波谱学(第二版)[color=#DC143C][font=黑体][size=4]附件在三楼----By阿宝[/size][/font][/color]
动物给药后取血浆。采用几种不同预处理方法处理血浆,比如有机溶剂沉淀,液液萃取,SPE柱等。然后进LC-MS进行代谢产物鉴定(注意是定性不是定量)。但是我如何证明哪种方法更好呢?据我所知只有基质效应,化合物数量来证明哪种方法更好。有没有其他的指标呢?发文章总不能说用肉眼看,感觉哪个好吧。有没有确定的指标来证明呢?感谢大牛的解答!!
老师:我这有一从红豆杉浸膏中提取的化合物,从色谱图保留时间看既不是10-DAB,也不是9-DHB ,会是何种物质呢? [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=49161]紫杉烷类化合物[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=49162]紫杉烷类化合物[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=49163]紫杉烷类化合物[/url]
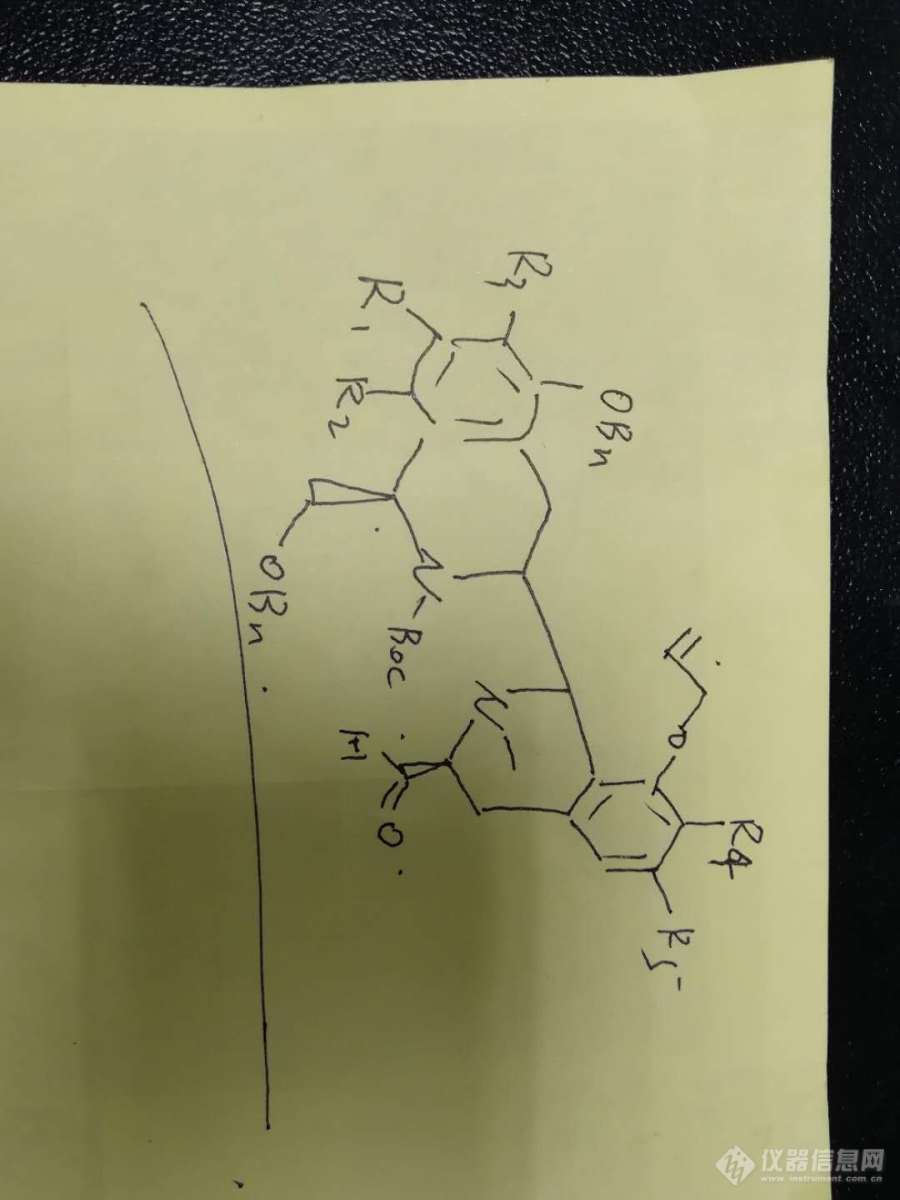
一个未知 的复杂化合物,查不到文献,以下为其大概的结构式,分子量约700多,以下尝试了几种方法、[img=,401,312]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131304270646_2129_3116636_3.png!w401x312.jpg[/img]刚开始用的溶剂是乙腈,体系为TFA乙腈体系,无法得到理想的峰形,而且尝试了很多种都不可以,后来尝试可一个梯度,这个梯度下出峰尖锐,但是并不理想,有峰未分离开来。(这个梯度是尝试了很多种后选择的分离度相对较好的条件。)梯度:0 50% 50% 5 50% 50% 9 15 % 85% 17 15% 85% 19 5% 95% 27 5% 95% 27.01 50% 50%后来尝试往有机相加了一定比例的甲醇,约30%(尝试过15-45%)基本差不多。乙腈溶解,0.1%TFA和30%+70%乙腈做流动相,梯度跟上图一样,出了两个峰,但实验员反映,打过核磁没有发现同分异构体也没有其余杂质。如下图[img=,690,345]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131309278474_7582_3116636_3.png!w690x345.jpg[/img]后来怀疑会跟流动相中的甲醇反应,于是将溶剂换成了甲醇,梯度流动相与上图一致,发现峰变成了一个,全部转化为左边峰,证明产品会与甲醇反应。(也试过异丙醇,一样会反应)如下图。[img=,690,353]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131312161097_8267_3116636_3.png!w690x353.jpg[/img]因此这个体系不可取了,实验员认为TFA可能也会与化合物反应,并且猜测化合物为中性,让用纯水,但是在我看来中性化合物的话,PH的影响对其在液相中的保留时间分离度不会有明显变化,但是还是尝试了一下纯水体系。乙腈溶解,纯水和乙腈作为流动相,梯度与上面相同,保留时间延后,分离度变好。但是左边依然有一个峰,这与最开始TFA和乙腈体系下有点相似,左边有一个未分离开来的小峰,在这个体系下分开了。那么说明这个化合物并非是中性化合物吗?如下图[img=,690,351]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131317512074_2363_3116636_3.png!w690x351.jpg[/img]为了证明左边的峰是否是与甲醇反应的峰一致,于是又进了一针甲醇溶解的,梯度进样量与上相同。结果变成了三个峰,第三个峰变小了,中间的峰变高,并且多了第一个峰……[img=,690,339]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131325256497_9234_3116636_3.png!w690x339.jpg[/img]就以上几种尝试多了很多无法想通的问题:1、这个化合物到底是偏中性还是碱性。2、在TFA体系下,甲醇溶解后进样只出一个峰,而在纯水体系下,却出了三个峰,那么是否说明起始在TFA的体系下也有三个峰,只是未能完全分开。3、谁做过类似的化合物,是否有较好的分析方法呢?最新的情况:还有一个化合物跟上面的相似,是其前一个步骤称为化合物1,只有一个基团不同,就是右下角是羟基,其余一样,上面那个就称为化合物2然后我们用上面的方法,TFA体系和纯水体系,有机相都为乙腈的条件下走了两针。在TFA体系下峰形良好,出峰时间约18.7,在中性体系下,峰形宽胖,出峰时间15.0min。然后呢还得知了其PKA值为6.7左右,上面那个化合物2PKA值5.3左右。那么为了更好的分离这两个物质,我选择的流动相应该PH在多少?已知TFA/磷酸这种较弱的PH下不可以分离,两个化合物峰靠的很近。在纯水体系下,两个化合物分离的很远,带羟基的化合物1出峰时间15.0min,但是峰形宽胖,带醛基的化合物2出峰时间27min,分离度良好,但是时间太靠后,试了好多其他梯度都没办法解决。我准备尝试0.1%的氨水体系,不知道是否可以。有没有其他更好的调节方式呢?望指教~~~
请教下化合物比如盐类像CsCl 等在峰的哪个位置,以及能从分子的结构解释这一规律吗
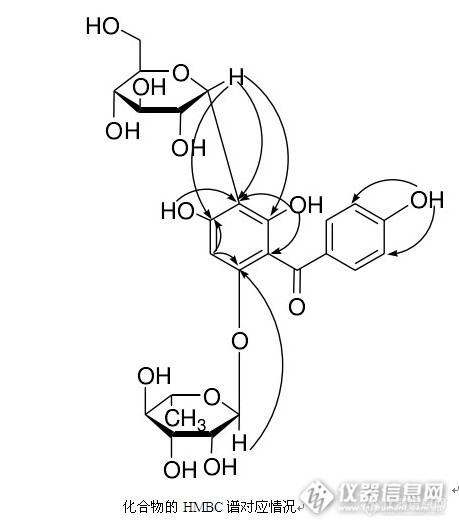
核磁共振波谱对查尔酮化合物的结构分析与鉴定 未知化合物为棕黄色粉末,mp 185~187℃。 D 20 :+11.0(c,0.1277,CH3OH);AlCl3反应呈阳性,提示该化合物为黄酮类化合物。 IR谱显示有酚羟基(3 333.5),羰基(1 604.9),碳氧键(1 282.2)http://ng1.17img.cn/bbsfiles/images/2014/11/201411281307_525037_2672081_3.jpgESI-MS谱给出的准分子离子(m/z):555.4+,577.4+,HRESI-MS给出+峰m/z:577.15139(计算值:577.15278),分子量为 554.16356,分子式为C25H30O14,不饱和度为 11。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281309_525038_2672081_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281309_525039_2672081_3.jpg13C-NMR谱中δ90~200 之间只有 11 个芳碳信号,其中有两组碳信号完全重合,应有 13 个碳,根据有关文献,推断可能为查尔酮类化合物,其中δ:192.9 应为羰基碳信号,说明该化合物含有两个苯环,一个羰基。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281309_525040_2672081_3.jpgTLC原位酸水解检出鼠李糖,在ESI-MS图谱中,有m/z为 409.4+和m/z为 391.4+存在,1H-NMR谱中,有δ5.09(1H,d,J=1.2Hz)和 4.65(1H,d,J=10.0Hz),δ1.05(3H,d,J=10.0Hz)说明该化合物连有两个糖,一个为鼠李糖,另一个为葡萄糖。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281310_525041_2672081_3.jpg根据以上信息,该化合物应含有两个苯环,一个羰基,一个鼠李糖,一个葡萄糖。1H-NMR谱中,δ8 以上有三个酚羟基宽质子单峰,10.23(1H,s),9.63(1H,s)和 8.61(1H,s),说明两个苯环上至少连有三个羟基。另外,在芳氢区显示一组AA′BB′系统的氢信号 7.55(2H,d,J=8.8Hz)和6.78(2H,d,J=8.4Hz),说明两个苯环中应有一对位取代的苯环,另外还有一单质子单峰6.29(1H,s),说明另一苯环上只有一个未被取代的芳氢,其它位置都被取代。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281311_525042_2672081_3.jpgDEPT谱表明含有 1 个甲基,1 个亚甲基,15 个次甲基,8 个季碳。与上述推测结果一致。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281311_525043_2672081_3.jpg通过HSQC和HMBC,对该化合物的碳、氢信号进行了归属http://ng1.17img.cn/bbsfiles/images/2014/11/201411281312_525044_2672081_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281312_525045_2672081_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281315_525046_2672081_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281315_525047_2672081_3.jpg结合HMBC谱发现,δ10.23羟基氢与δ115.0(3′、5′位碳)远程相关,证明此羟基连在 4′位碳上。δ8.61 羟基氢与δ155.4(6 位碳)和δ106.2(5 位碳)及δ109.4(1 位碳),远程相关,证明此羟基连在6 位碳上。δ9.63 羟基氢与δ157.8(4 位碳)和δ106.2(5 位碳)远程相关,证明此羟基连在4位碳上。δ5.09(鼠李糖端基氢)与δ154.6(2 位碳)远程相关,证明鼠李糖连在 2位碳上。δ4.65(葡萄糖端基氢)与δ106.2(5 位碳)和δ[/f







 我要推广仪器
我要推广仪器
 下载APP
下载APP