我想分析芳烃中微量的甲醇,色谱仪带FID检测器,望高手详细告诉我方法、 柱子(填充柱)、色谱条件,注意事项,谢谢!
[color=#444444]现有C6-C8烃类混合物,其中芳烃含量500ppm-20ppm之间,相对芳烃含量准确定量,应该选择什么方法呢?[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]内标分析能准确么?紫外分光光度计呢?[/color]
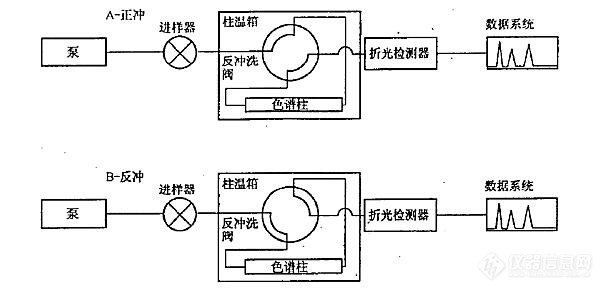
[b][font=宋体]问题描述:使用[/font]SH 0806-2008[font=宋体]标准方法测定柴油和馏程范围为[/font]150~400[font=宋体]℃[/font][font=宋体]的石油馏分中单环芳烃、双环芳烃、三环以上芳烃和多环芳烃含量,有哪些注意事项?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])系统结构原理如图[/font]6-41[font=宋体]所示,系统使用正庚烷做流动相,单输送泵、极性色谱柱(氨基或者氰基柱)、四通阀和示差检测器实现分离[/font].[align=center][img=,605,292]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047269515_6030_3389662_3.jpg!w605x292.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-41 SH 0806 [font=宋体]方法的硬件原理[/font][/i][/align][font=宋体]([/font]2[font=宋体])待机状态和进样状态下(即状态[/font]A[font=宋体]),流动相自右向左流过极性色谱柱,柴油中的烃类和芳烃类组分实现分离。理想情况下,单环芳烃、双环芳烃、多环芳烃依次在色谱柱出口流出,如图[/font]6-42[font=宋体]所示。[/font][align=center][img=,508,274]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047383890_9606_3389662_3.jpg!w508x274.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-42 [font=宋体]理想的色谱柱内样品分离谱图[/font][/i][/align][font=宋体]([/font]3[font=宋体])当双环芳烃流出色谱柱后(色谱图中[/font]A[font=宋体]点位置,或称为切换点),四通阀旋转,系统状态变为反吹。色谱柱内流动相的方向变成自左至右,将三环以及三环以上的芳烃类物质反吹出色谱柱,在示差检测器上表现为单峰,如图[/font]6-43[font=宋体]所示。[/font][align=center][img=,536,271]https://ng1.17img.cn/bbsfiles/images/2021/03/202103241047483421_9431_3389662_3.jpg!w536x271.jpg[/img][/align][align=center][i][font=宋体]图[/font]6-43[font=宋体]最终样品分离谱图[/font][/i][/align][font=宋体]([/font]4[font=宋体])该方法的原理是比较理想化的,干扰因素也比较多。柴油中的单环芳烃、双环芳烃、多环芳烃是否可以清晰彻底的分离开,是难以保证的。况且柴油中的二烯烃、杂环类、酯类化合物等都会对分析结果带来影响。[/font][font=宋体]([/font]4[font=宋体])色谱柱的选择十分重要,具体的选型需要咨询色谱柱厂家。[/font][font=宋体]([/font]5[font=宋体])切换点的选择非常重要。样品组成可能比较复杂,[/font]A[font=宋体]点可以选择的时间窗口就会比较窄,需要多次重复实验寻找合适的切换点。分析条件需要非常稳定,需要较为严格的控制流动相组成、泵输送流速以及色谱柱温度,以免影响保留的重复性。[/font][font=宋体]([/font]6[font=宋体])需要注意流动相不稳定导致保留时间漂移:[/font][font=宋体]保留时间的漂移是最为常见的问题。在进样系统性能测试标准样品时,芳烃的保留时间长时间的漂移,致使难以确定切换点。往往会耗费较多时间来平衡系统,等待保留时间稳定,从而降低分析效率。其本质的原因在于流动相的不稳定。[/font]SH 0806-2008[font=宋体]系统分离原理属于正相液相色谱,我们知道正相[/font]HPLC[font=宋体]一般不太容易得到良好的保留时间重复性。原因是在正相[/font]HPLC[font=宋体]分析中,流动相中的微量水会显著的改变其极性。假设流动相原先的极性为[/font]0.01[font=宋体],吸收微量水之后极性变为[/font]0.02[font=宋体],看上去似乎变化不大,但其实极性增大了一倍。尤其是使用硅胶色谱柱的场合,流动相与环境空气中的水蒸气发生交换,改变了极性,从而影响保留时间。在使用硅胶柱分析时,一般要避免使用彻底干燥的正己烷流动相,避免吸水造成保留不稳定,甚至需要特意在流动相中加入微量的水。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font][color=red] [/color]
如上:公司要分析芳烃溶剂油中的芳烃含量,按国标需用荧光指示剂吸附法,但是操作太麻烦。我尝试使用PONA软件和柱子进行分析,不知道可行性如何?有什么需要注意的地方?望高手指教。
有谁用GCMS检测过芳烃溶剂油中芳烃,烯烃烷烃含量的,沸点在168度--312度之间。想请教!
近期,德国在对消费品安全要求进行审查后,建议在GS(安全性认证(安全测试))控制列表中增加两种多环芳烃(PAH)致癌物。这将使GS 标志列表管控的多环芳烃总数达到18个。自愿性的德国GS标志认证基于美国环境保护署提出的技术指标列表对适用产品中的多环芳烃加以管控。目前,这份列表包含的 16种多环芳烃被分类为致癌物质。不过,最近由欧洲食品安全局(EFSA)和德意志研究联合会(DFG)参议院委员会认定的两种剧毒多环芳烃却未被列入表内。这两种物质——苯并荧蒽,苯并苝,都被列为第二类致癌物质,并被列入REACH法规附录XVII(第50项)。因此,德国倡议进一步控制消费品中的多环芳烃。这两种物质的加入将确保欧盟列出的所有八个第二类高致癌多环芳烃可通过GS标志进行管控。德国技术设备及消费品委员会(AtAV委员会)建议,申请GS标志应基于以下更新物质列表。在测试范围更新之后,ZEK 01.2-08规定的分析过程将是应用于GS标志认证的测试方法。经验交流中心(ZEK)第67次会议在德国德国技术设备及消费品委员会(ZEK-04-11号文件)的建议下同意了此次列表的扩大。从现在起,经过六个月的过渡期后,新的物质列表将应用于GS标志认证。
请问在北京哪里能分析渣油中重芳烃的成分,如苯并芘的含量,如何收费?
便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]结合固相微萃取装置检测土壤中8种多环芳烃方法概述 环境中的多环芳烃(PAHs)由有机物(如煤、石油和木材等)燃烧不完全而产生,是常见的环境和食品污染物。由于PAHs具有致癌、致畸和致突变性,更具有较强的持久性,美国环保署已把16种多环芳烃列入优先控制有毒有机污染物黑名单中,在我国环保部第一批公布的68种优先污染物中,PAHs有7种。根据《全国土壤污染状况调查公报》,全国土壤总的超标率为16.1%,总体状况不容乐观,其中有机污染物以六六六、滴滴涕和多环芳烃为主,多环芳烃的点位超标率达到1.4%,仅次于滴滴涕。在不同类型用地中,耕地是多环芳烃的主要污染区,在典型地块的周边土壤污染调查中,结果表明工业废弃地、工业园区、采油区、采矿区、污水灌溉区及干线公路两侧都是多环芳烃的主要污染地块,在调查的同地块中超标点位分别占34.9%、29.4%、23.6%、33.4%、26.4%和20.3%。由此可见,建立现场快速分析土壤中多环芳烃的分析方法,判断污染程度,对保护人体健康具有重要的实际意义。 土壤基体复杂,且PAHs浓度低(痕量或超痕量),难以直接测定,必须采用一定的预处理技术使其可以达到可检测的水平。对于PAHs的检测大多采用GC、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]或LC方法,便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]技术是传统的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]技术的衍生和发展,作为现场快速检测设备,更真实地反映了污染物的排放情况,而固相微萃取是集采样,浓缩,萃取及进样于一体的无需使用溶剂的一种前处理方法,操作方便、简单,省时省力,将其与体积小、重量轻及分析速度快的Mars-400 Plus便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]相结合,能及时快速地应对一些突发事故。 因此本文采取选用SPME方法结合Mars-400 Plus便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]检测土壤中的PAHs,建立了便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]检测土壤中的萘、苊烯和苊等8种多环芳烃的分析方法。主要仪器与试剂 2.1 仪器 Mars-400 Plus便携式[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪; LTM DB-5ms 快速[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱(5 m×0.1 mm×0.4 μm); SPME综合前处理装置:聚光科技; 便携式分析天平。 2.2 试剂 标准样品:浓度2000 mg/L的8种多环芳烃标样 2.3 材料 微量[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url](10 μL); 微量[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url](100 μL); 微量[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url](1000 μL); SPME手柄:购于Supelco公司; PDMS/DVB萃取头:65μm,部分交联,蓝色平头; 涡旋振荡器; 载气:氦气,纯度99.999%。标准样品配制 3.1 标准样品储备液配制 取50 μL 8种多环芳烃的有机物母液于5 ml容量瓶中,以乙腈为溶剂,定容,得到20mg/L的标准样品储备液。 3.2 标准系列样品溶液的配制 取5个40 mL的样品瓶,分别称取5 g土壤,向各样品瓶中分别加入0.5 μL、1 μL、2 μL、5 μL、10 μL的标准样品储备液,再分别加入20 mL水萃取溶剂,配制成目标化合物浓度分别为0.5 μg/L 、1 μg/L、2 μg/L、 5 μg/L、10 μg/L的标准系列样品溶液,放入搅拌子,用涡旋振荡仪振荡5min,密封待测。利用固相微萃取技术,由低浓度到高浓度依次萃取和分析,绘制校准曲线。样品分析 4.1 样品分析条件表 1 样品分析条件[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571978840921334.png[/img] 4.2 样品分析步骤 (1)打开Mars-400 Plus便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url],并调试稳定; (2)设定SPME综合前处理装置的分析条件,将标准样品溶液放入SPME综合前处理装置的加热模块中,预热和平衡样品; (3)将SPME萃取纤维插入老化口,点击装置上的老化按钮,老化纤维2min; (4)取样品溶液,放入样品瓶座中平衡; (5)待样品溶液平衡完成,纤维老化完成后,将SPME插入样品瓶中,使萃取纤维置于液面以上位置,点击萃取界面的启动按钮,此时磁力搅拌器启动,进行样品搅拌萃取; (6)打开主机的选择方法界面,选择固相微萃取进样方法,设定分析条件,激活方法,萃取完成后,将SPME手柄插入进样口,运行方法; (7)按照步骤(2)至(5)从低到高分析标准系列样品溶液,建立校准曲线。当样品高低浓度交叉分析时,需在中间插入样品空白分析,降低残留影响; (8)标准样品分析完成后输出分析报告,并进行仪器维护。5 结果与讨论 5.1 绘制校准曲线 按照章节4.2的样品分析方法,从浓度低到浓度高分析标准系列样品。本试验采用特征离子定量法进行定量。以样品浓度作为横坐标,以样品特征离子峰面积作为纵坐标,绘制校准曲线(图 1,表 2)。[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571978879231149.png[/img]图 1 土中8种PAHs总离子流图 注:1-萘;2-苊烯;3-苊;4-芴;5-菲;6-蒽;7-荧蒽;8-嵌二萘表 2 土中8种PAHs的校准曲线[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571978916876639.png[/img]表 3 土中8种PAHs在浓度为1 μg/L的精密度分析结果 注:标准溶液以干净土壤为基质。 图 1是结合SPME进行萃取后,便携式[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]分析得到的8种PAHs的总离子流图。表 2为8种PAHs的标准曲线,由表 2可以看出,8种PAHs的标准曲线线性较好,相关系数(R)都在0.99以上。 5.2 方法精密度和准确度 分别采用低浓度1 μg/L和高浓度5 μg/L的8种多环芳烃标准溶液进行6组平行对照试验,计算其相对标准偏差,得到方法的精密度。以土壤为基质,用5 μg/L的8种多环芳烃标准溶液进行6组平行对照试验,计算加标回收率,从而得到方法的准确度,加标回收率应在70%~130%之间。表 3 土中8种PAHs在浓度为1 μg/L的精密度分析结果[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571978969658687.png[/img] 注:样品溶液以干净土壤为基质。表 4 土中8种PAHs在浓度为5μg/L的精密度分析结果[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571978992146391.png[/img] 注:样品溶液以干净土壤为基质。 如表 3所示,在浓度为1 μg/L时,6组平行试验得到8种PAHs的相对标准偏差(RSD)在20%以内。如表 4所示,在浓度为5μg/L时,6组平行试验得到8种PAHs的相对标准偏差(RSD)在17%以内,精密度良好。表 5 土中8种PAHs在浓度为5μg/L的加标回收率[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571979013455655.png[/img] 由表5可知,8种PAHs的加标回收率都在70%~130%之间,方法准确度较好。 5.3 方法检出限 检出限采用0.5 μg/L的标准样品在设定条件下进样分析,计算每种物质对应的信噪比,以3倍信噪比(S/N)计算。表 6 结合SPME分析土中8种PAHs的方法检出限[img=image.png]https://i4.antpedia.com/attachments/att/image/20191025/1571979025491803.png[/img] 如表 6所示,8种PAHs的检出限在0.185~0.383 μg/L之间。总结 从实验结果看来,采用SPME技术结合便携[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]分析土中多环芳烃的方法是有效可行的。结果表明,土中8种多环芳烃的相关系数都达到了0.99,且在低浓度和高浓度时,8种多环芳烃的RSD都在20%以内,重复性结果较好。从检出限上看,8种多环芳烃的检出限也在0.185~0.383 μg/L之间。因此,利用SPME结合便携[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]的方法操作简单、分析速度快,是作为现场快速分析行之有效的分析方法之一,可及时应对水、土污染等突发事故
大家好,我想用自己制备的层析柱,分离煤液化油中的饱和分(饱和烃)、芳香分(芳香烃)和胶质(极性芳香烃),然后对得到的芳香分用C18柱进行分析。但是我怕我制备的芳香分不纯,可能会含有微量的饱和烃,这部分微凉的芳香烃可能会对C18柱造成污染。所以想请教用什么简便的方法能除去芳香烃(不是极性芳香烃)中微量的饱和烃,谢谢。如果用固相萃取柱,用哪种好些,另外溶剂该怎么选?谢谢大家。
在芳烃含量测量试验中,苯是有极性的,对吗?
GS标志物质列表中将新增两种多环芳烃 来源:技术壁垒资源网 时间:2011-11-09近期,德国在对消费品安全要求进行审查后,建议在GS安全性认证(安全测试)控制列表中增加两种多环芳烃(PAHs)致癌物质。这将使 GS 标志列表管控的多环芳烃总数达到 18个。 自愿性的德国GS标志认证基于美国环境保护署提出的技术指标列表对适用产品中的多环芳烃加以管控。目前,这份列表包含的 16种多环芳烃被分类为致癌物质。不过,最近由欧洲食品安全局(EFSA)和德意志研究联合会(DFG)参议院委员会认定的两种剧毒多环芳烃却未被列入表内。这两种物质——苯并荧蒽,苯并苝,都被列为第二类致癌物质,并被列入REACH法规附录XVII(第50项)。因此,德国倡议进一步控制消费品中的多环芳烃。这两种物质的加入将确保欧盟列出的所有八个第二类高致癌多环芳烃可通过GS标志进行管控。 德国技术设备及消费品委员会(AtAV 委员会)建议,申请GS标志应基于以下更新物质列表。在测试范围更新之后,ZEK 01.2-08规定的分析过程将是应用于GS标志认证的测试方法。 经验交流中心(ZEK)第67次会议在德国德国技术设备及消费品委员会(ZEK-04-11 号文件)的建议下同意了此次列表的扩大。从现在起,经过六个月的过渡期后,新的物质列表将应用于 GS 标志认证。如有疑问,请致电PONY谱尼测试零七五五二六零五零九零九
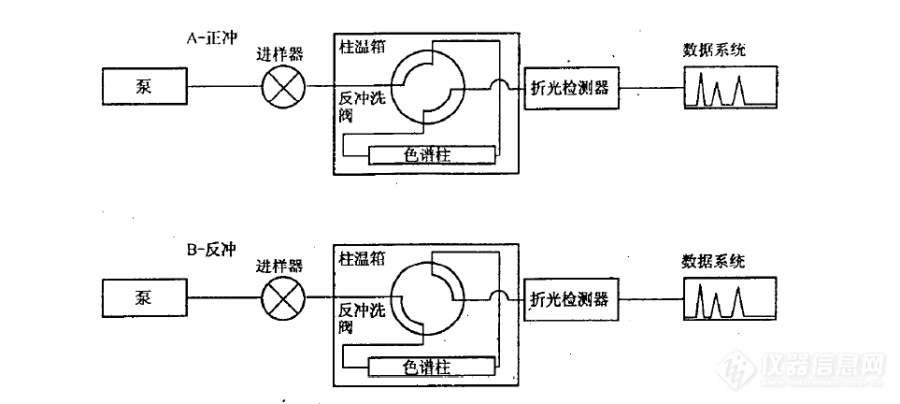
[align=center][font=宋体][size=14.0000pt]SH 0806-2008 [font=宋体]中间馏分芳烃含量的测定 分析方法的注意事项[/font][font=Calibri]----[/font][font=宋体]流动相[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]概述:[/font]SH0806-2008[font=宋体]标准使用高效液相色谱方法测定柴油和馏程范围为[/font][font=Calibri]150-400[/font][font=宋体]℃的石油馏分中单环芳烃、双环芳烃、三环以上芳烃和多环芳烃含量。该分析方法虽然比较简单,但是分析条件的控制比较重要。[/font][/size][/font][font=宋体][size=12.0000pt]在实验操作中,尤其需要注意流动相的问题,流动相不良会造成保留时间漂移从而使得色谱柱切换时间选择发生困难。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]一[/font] [font=宋体]原理介绍[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]系统结构原理如图[/font]1[font=宋体]所示,系统使用正庚烷做流动相,单输送泵、极性色谱柱(氨基或者氰基柱)、四通阀和示差检测器实现分离。[/font][/size][/font][font=宋体][size=12.0000pt][font=宋体]待机状态和进样状态下(即状态[/font]A[font=宋体]),流动相自右向左流过极性色谱柱,柴油中的烃类和芳烃类组分实现分离。理想情况下,单环芳烃、双环芳烃、多环芳烃依次在色谱柱出口流出,如图[/font][font=Calibri]2[/font][font=宋体]所示。[/font][/size][/font][font=Calibri][size=12.0000pt][img=,690,309]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010841562718_6781_1604036_3.png!w690x309.jpg[/img] [/size][/font][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]1 SH 0806 [font=宋体]方法的硬件原理图[/font][/size][/font][/align][align=center][font=Calibri][size=12.0000pt][img=,690,348]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010842112720_7230_1604036_3.png!w690x348.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]2 [font=宋体]色谱柱内样品理想分离示意图[/font][/size][/font][/align][font=宋体][size=12.0000pt][font=宋体]当双环芳烃流出色谱柱后(色谱图中[/font]A[font=宋体]点位置,或称为切换点),四通阀旋转,系统状态变为反吹。色谱柱内流动相的方向变成自左至右,将三环以及三环以上的芳烃类物质反吹出色谱柱,在示差检测器上表现为单峰,如图[/font][font=Calibri]3[/font][font=宋体]所示。[/font][/size][/font][align=center][font=Calibri][size=12.0000pt][img=,690,351]https://ng1.17img.cn/bbsfiles/images/2020/06/202006010842234040_3244_1604036_3.png!w690x351.jpg[/img] [/size][/font][/align][align=center][font=宋体][size=9.0000pt][font=宋体]图[/font]3 [font=宋体]最终谱图[/font][/size][/font][/align][align=center][font=宋体][size=12.0000pt][font=宋体]二[/font] [font=宋体]分析注意要点[/font][/size][/font][/align][font=宋体][size=12.0000pt]首先要注意该方法的原理是比较理想化的,干扰因素也比较多。柴油中的单环芳烃、双环芳烃、多环芳烃是否可以清晰彻底的分离开,是难以保证的。况且柴油中的二烯烃、杂环类、酯类化合物等都会对分析结果带来影响。[/size][/font][font=宋体][size=12.0000pt]其次,色谱柱的选择十分重要,具体的选型需要咨询色谱柱厂家。[/size][/font][font=宋体][size=12.0000pt]再次,切换点的选择非常重要。[/size][/font][font=宋体][size=12.0000pt][font=宋体]样品组成可能比较复杂,[/font]A[font=宋体]点可以选择的时间窗口就会比较窄,需要多次重复实验寻找合适的切换点。[/font][/size][/font][font=宋体][size=12.0000pt]分析条件需要非常稳定,需要较为严格的控制流动相组成、泵输送流速以及色谱柱温度,以免影响保留的重复性。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]三[/font] [font=宋体]常见问题[/font]——流动相[/size][/font][/align][font=宋体][size=12.0000pt]保留时间的漂移是最为常见的问题。在进样系统性能测试标准样品时,芳烃的保留时间长时间的漂移,致使难以确定切换点。往往会耗费较多时间来平衡系统,等待保留时间稳定,从而降低分析效率。[/size][/font][font=宋体][size=12.0000pt]其本质的原因在于流动相的不稳定。[/size][/font][font=宋体][size=12.0000pt][font=宋体]笔者曾经在使用该系统时,长时间重新系统后,连续进样[/font]10[font=宋体]余次系统测试标样,发现芳烃的保留时间不断发生缩短,认为色谱柱未彻底平衡。[/font][/size][/font][font=宋体][size=12.0000pt][font=宋体]第二天更换了流动相(新换的流动相没有彻底封口放置在实验室[/font]10[font=宋体]小时),进样[/font][font=Calibri]3[/font][font=宋体]次,芳烃的保留时间较为稳定。[/font][/size][/font][font=宋体][size=12.0000pt]后来考虑了一下,原因应当为新换的正庚烷中水含量已经与空气中的水含量交换平衡,或者说水含量已经比较稳定,进而使得芳烃保留稳定。[/size][/font][align=center][font=宋体][size=12.0000pt][font=宋体]四[/font] [font=宋体]小结[/font][/size][/font][/align][font=宋体][size=12.0000pt]SH 0806-2008[font=宋体]系统分离原理属于正相液相色谱,我们知道正相[/font][font=Calibri]HPLC[/font][font=宋体]一般不太容易得到良好的保留时间重复性。原因是在正相[/font][font=Calibri]HPLC[/font][font=宋体]分析中,流动相中的微量水会显著的改变其极性。假设流动相原先的极性为[/font][font=Calibri]0.01[/font][font=宋体],吸收微量水之后极性变为[/font][font=Calibri]0.02[/font][font=宋体],看上去似乎变化不大,但其实极性增大了一倍。[/font][/size][/font][font=宋体][size=12.0000pt]尤其是使用硅胶色谱柱的场合,流动相与环境空气中的水蒸气发生交换,改变了极性,从而影响保留时间。在使用硅胶柱分析时,一般要避免使用彻底干燥的正己烷流动相,避免吸水造成保留不稳定,甚至需要特意在流动相中加入微量的水。[/size][/font]
[font=微软雅黑]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-4153.html[/url]多环芳香烃(PolycyclicAromatic Hydrocarbons,简称PAH或PAHs)又称多环性芳香化合物或多环芳香族碳氢化合物,是芳香族碳氢化合物的一种特例,由不包含杂环或取代基的芳香环所组成;其致癌、光致毒、破坏免疫系统等对人体造成巨大危害。[/font][font=微软雅黑]首先,多环芳烃最明显的危害来至于其致癌性,多环芳烃的致癌性可能是因其易于和细胞内的DNA、RNA等遗传物质结合而起致癌作用。[/font][font=微软雅黑]其次,多环芳烃更危险在于它们暴露于太阳光中紫外线辐射时的光致毒效应。太阳光中可见光区和紫外光区的光对多环芳烃的毒性有显著影响。[/font][font=微软雅黑]最后,多环芳烃可以引起机体的免疫抑制反应,表现为血清免疫学指标的改变。[/font][font=微软雅黑]鉴科检测参考《HJ 784-2016 土壤和沉积物多环芳烃的测定高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]》,建立了利用全自动固相萃取仪(Fotector Plus)结合高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]检测沉积物中多环芳烃残留量的方法。在100mL丙酮-正己烷(1+1)提取后,使用Auto EVA-08IR浓缩至1mL后 Fotector Plus全自动固相萃取仪净化,自动完成 SPE 柱活化、样品上样、淋洗、收集等步骤,收集液再氮吹浓缩、溶剂转换、定容后,用HPLC检测。[/font][font=微软雅黑][/font]
方法采用月旭ULTIMATE PAH专用柱分离,并与其他分离柱比较,其分离度在含油较高的样品效果最好。 【第二届网络原创参赛作品】微波萃取测定烤肉中的多环芳烃 郑建明 多环芳烃(Polycyclic Aromatic Hydrocarbons)是环境中常见的污染物之一,其来源于有机物热解和不完全燃烧,因此熏烤类食品在制作过程中的熏烟中含有较高含量的多环芳烃。烤制时,滴于火上的食物脂肪焦化产物发生热聚合反应,形成多环芳烃类化合物,附着于食物表面,这是烤制食物中多环芳烃类的主要来源。另外,食物烤焦烤糊时,脂肪因高温裂解,产生自由基,并相互结合,即发生热聚合反应,由此生成多环芳烃物质。长期食用含有多环芳烃的食物对健康将产生潜在威胁。多环芳烃是指由2个或2个以上苯环以线状角状或簇状排列的化合物,是一种广泛存在于环境,食品及生物体内的污染物。其中16种多环芳烃因具有致畸,致癌和致突变的作用而被美国环保署列为最具危害的有机污染物。本文为低含量多环芳烃,高脂肪,高蛋白的熏烤肉质样品的测定提供借鉴。1 实验部分1.1 仪器与试剂1.1.1液相色谱仪 LC-310配紫外检测器(天瑞仪器股份有限公司生产),色谱柱:LC-PAH色谱柱,250mm×4.6mm×5.0um,柱温:30℃,流动相及流速:50%乙腈+50%水 1.5 mL/min,检测波长:220 nm,进样量:10 μL。1.1.2 微波系统MSD-8 上海新仪微波设备有限公司1.1.3 加热板 上海新仪微波设备有限公司1.1.4试剂正己烷 色谱纯 天津市永大化学试剂开发中心丙酮 色谱纯 无锡市展望化工试剂有限公司二氯甲烷 色谱纯 天津市大茂化学试剂厂硫酸 优级纯 无锡市展望化工试剂有限公司多环芳烃混标100 mg/L 百灵威化学品有限公司1.2 样品处理准确称取2.0 g样品于特氟龙杯中,加入30 mL(1:1)丙酮和正己烷的混合溶液,放入微波萃取系统中,升温至100℃,保持20 min,冷却至室温,将萃取罐体打开放在加热板上加热加热挥发至10 mL左右,加5 mL硫酸进行脱脂(1~2次),离心取上层溶液通过活化的硅胶/无水硫酸钠(湿式过柱法,以正己烷为载体 1mL无水硫酸钠、10mL硅胶、1mL无水硫酸钠)层析柱后,加入10mL(1:1)二氯甲烷-正己烷淋洗多环芳烃组分,收集淋洗液,浓缩定容至5 mL,并用液相色谱外标法进行定量分析。2 结果与讨论2.1 标准溶液配制用移液器分别吸取0.25、0.5、1.0 、2.5、5 mL浓度为100 mg/L的多环芳烃标准品于50 mL容量瓶中,用丙酮和正己烷(1:1)的混合溶液定容并摇匀,即得到浓度分别为0.5、1.0、2.0、5.0、10.0 mg/L多环芳烃混标溶液。2.2 标准曲线与方法检出限分别吸取10 μL浓度为0.0、0.5、1.0、2.0、5.0、10.0 mg/L多环芳烃混标溶液进样,以多环芳烃各种物质的峰面积为纵坐标,以标液浓度为横坐标绘制工作曲线。多环芳烃的标准曲线和方法检出限见表1.由表1可见,本方法对测定物质具有较好的线性和较低的检出限。============================================下载地址: http://www.instrument.com.cn/bbs/images/affix.gif微波萃取测定烤肉中的多环芳烃.doc============================================
我要测定石油中多环芳烃的含量,这个我用标准SN-T 1877.3-2007矿石油中多环芳香烃的测定可以吗?也没有找到任何关于石油中PAHs测定的资料,希望大家赐教
去年做过16中多环芳烃,分离度也不错。今天调用之前的方法标准品(10ug/ml)出的十几个峰特别小,因为时间关系没来的及做高浓度的,标准品是二氯甲烷中的多环芳烃,我拿已睛配置的浓度点,想知道这样有没有影响?剃度是甲醇水,紫外检测器。有做过多环芳烃的大侠可以指点一下吗?万分感激!!!
小弟最近遇到要检测芳烃油中多环芳烃的检测,参考文献,前处理过程主要如下:1.样品用环己烷(10 mL)稀释2.用8 mL DMSO萃取环己烷两次3.DMSO层用NaCl溶液稀释后,再用环己烷萃取4.环己烷吹干后,正己烷复溶,固相萃取柱净化我的问题在于,遇到一个样品,环己烷稀释后用DMSO萃取时,DMSO与环己烷不能完全分层,离心的话中间会有浮渣。也试过环己烷稀释后离心,取上清再用DMSO萃取,但是情况也没有改善。求教各位大神如何能解决DMSO与环己烷不能分层的问题~
方法名称:海水中16种多环芳烃的测定 气相色谱-质谱法 疑问:1、为什么选择玻璃纤维膜进行水样的过滤,不能采用其他的膜?标准号:GB/T 26411—2010 2、C18(500mg/3mL)柱的吸附PAHs效果如何(柱子的含碳量是选择17% 还是 11%),回收率能否达到60%以上?仪器设备:赛默飞GC-MS/MS 3、方法是1 L水样过柱, 如何把1 L水样导入C18柱子中,如果采用分液漏斗 那么分液漏斗1 L非常巨大,SPE装置上面最多能放四个,估计三个耗费 大量的时间精力;如果采用SPE大体积采样管能达到很好的效果但是费用 很高一根管240元。所以想请问各位有什么好的处理方法。试实验流程:按照标准方法进行(附件方法) 4、此方法中已经选用替代物(多环芳烃的五种替代物)作为内标,为什么还 要选择内标溶液(四氯间二甲苯或者六甲基苯),内标溶液的作用在哪? 5、本实验中还有那些注意事项(例如SPE的使用、柱子的活化、水样过柱的 速率等),需要提前准备和想到,请各位指点。大家共享。
实验论文:微波萃取测定烤肉中的多环芳烃
1.PAHs是什么?http://www.liupingnian.cn/wp-content/uploads/2014/06/PAH1.pnghttp://www.liupingnian.cn/wp-content/uploads/2014/06/PAH2.pnghttp://www.liupingnian.cn/wp-content/uploads/2014/06/PAH3.png2.PAHs哪里来?食品中的PAH污染有不同的来源,其中最主要的是环境和食品加工过程的污染。加工过程又被认为是造成食品PAH污染的最主要方式,包括食品的烟熏、烘干、烹饪过程。加工过程的污染的例子:A. 沥青路面晾晒谷物,大豆及各种食品(在中国大部分地区都存在,国外未知)B. 机动车路边等室外晾晒食品(大气中的PAHs吸附在食品上,特别是容易出油的肉,鱼等)C.熏鱼,熏肉等传统加工方法(确实是用烟熏的,现代的加工法中有些只是采用烟熏液,可以显著降低PAHs的污染)D.溶剂提取方式提取食用油(在油桶上的加工方式为浸提),而溶剂会有含有PAHs的风险。E. 厨房油烟,油温太高时,也容易产生多环芳烃。F. 烧烤食品3.PAHs的危害2002年在欧洲,食品科学委员会(SCF)已对33种PAH进行了评价。评价的结果显示在33种PAH中有15种物质在实验动物的体内实验中证明对体细胞具有致突变性和基因毒性,这15种物质分别为苯并蒽,苯并-, -, 荧蒽,苯并二萘嵌苯(benzoperylene),苯并芘,屈(chrysene),环戊芘(cyclopentapyrene), 二苯并蒽(dibenz anthracene),二苯并-,二苯并-,二苯并-,二苯并芘,茚并芘和5-甲基屈。许多具有毒性作用的PAH会在凹陷的区域结合形成复合物。动物实验表明PAH具有多种毒性作用,如血液毒性、生殖和发育毒性及免疫毒性。在低计量时就具有致癌性和致畸性。具有致癌性和致畸性的PAH分子量通常比较大,含有4个或4个以上的环。对于许多PAH,它的潜在的致癌性造成了主要的危害和危险性。已经证实在燃烧过程中产生的许多PAH、煤焦油和包括PAH的各种混合物在实验动物体内、体外的基因毒性和诱变性实验中都显示了致癌性。总的来说,许多证据表明基因毒性与在PAH暴露下DNA加和物形成、突变和癌症发生的致癌机制相似。对实验动物的各种检验结果显示,15种具有基因毒性的PAH,除了苯并 二萘嵌苯外,其余均具有显著的致癌性。虽然只有苯并芘可以使用膳食管理的方法检测,但其他化学物也均被认为对人类具有潜在的基因毒性和致癌性。由于通过饮食摄入PAH对健康有长期副作用,因此将这些化合物列为优先评估的项目进行危险性评估。4. PAHs的限量因为其毒性太大,制定限量的趋势是接近现代最先进仪器的检出限和接近自然本底值。5.PAHs的测试标准中国标准: CJ/T 147-2001城市供水多环芳烃的测定液相色谱法 CNS 15169-2007香品燃烧所产生之多环芳香烃化合物测定法 CNS 15289-2009硫化橡胶制品中加工油多环芳香烃之测定法 CNS 16000-12-2011室内空气-第12部:多氯联苯(PCBs)、多氯二苯并对戴奥辛(PCDDs)、多氯二苯并呋喃(PCDFs)及多环芳香烃(PAHs)之采样策略 GB/T 23213-2008植物油中多环芳烃的测定气相色谱-质谱法 GB/T 24893-2010动植物油脂多环芳烃的测定 GB/T 26411-2010海水中16种多环芳烃的测定气相色谱-质谱法 GB/T 28189-2011纺织品多环芳烃的测定 GB/T 29614-2013硫化橡胶中多环芳烃含量的测定 GB/T 29616-2013热塑性弹性体多环芳烃的测定气相色谱-质谱法 GB/T 29670-2013化妆品中萘、苯并蒽等9种多环芳烃的测定气相色谱-质谱法 GB/T 29784.1-2013电子电气产品中多环芳烃的测定第1部分:高效液相色谱法 GB/T 29784.2-2013电子电气产品中多环芳烃的测定第2部分:气相色谱-质谱法 GB/T 29784.3-2013电子电气产品中多环芳烃的测定第3部分:液相色谱-质谱法 GB/T 29784.4-2013电子电气产品中多环芳烃的测定第4部分:气相色谱法 GBZ/T 160.44-2004工作场所空气有毒物质测定多环芳香烃类化合物 HJ 478-2009水质多环芳烃的测定液液萃取和固相萃取高效液相色谱法 HJ 646-2013环境空气和废气气相和颗粒物中多环芳烃的测定气相色谱-质谱法 HJ 647-2013环境空气和废气气相和颗粒物中多环芳烃的测定高效液相色谱法[table=100%,tra
本文建立了柴油中多环芳烃的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱([url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url])分析方法。柴油的成分比较复杂,如果不经过前处理过程,很难检测到其中的多环芳烃;但是在经过氟罗里硅土柱的净化并浓缩之后,就能实现多环芳烃的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]检测。该方法操作简单,有望应用于柴油中多环芳烃的检测。柴油中碳氢化合物的主要成分为正烷类、含支链的烷类及环烷类、异戊二烯化合物和芳香族类等四类,其中含量比例最高的为正烷类,其次为支烷、环烷类及芳香族类,而最低的则为异烷类。依毒性来说,以芳香族最毒,而芳香族中包含苯、甲苯、二甲苯以及多环芳香族碳氢化合物等都具有毒性或致癌性。当前,柴油被广泛地应用于各种工业生产活动中,这种广泛的应用使柴油的污染带来的问题变得日益严峻。所以建立柴油中多环芳烃的分析方法是非常必要的。[b]实验部分[/b]仪器岛津公司[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-QP2010 Plus样品前处理先用正己烷10ml预淋洗氟罗里硅土(Florisil)柱,再加入0.5ml柴油,并采用20ml二氯甲烷与正己烷的混合液(体积比1:1)洗脱。将得到的洗脱液在N2下浓缩至约0.5ml后在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]上进样分析。[img=,280,519]http://img.vogel.com.cn/2011/20100705/2142400.jpg[/img][b]分析条件[/b]进样口:260℃;柱温:90℃(1min)8℃/min180℃15℃/min 280℃(12min);进样方式:不分流;载气:氦气;色谱柱:RTX-5ms(30m×0.25mm×0.25um);载气柱流量:1.79ml/min;离子源:200℃;进样量:1μl;电离方式:EI;数据采集模式:SIM。[img=,553,719]http://img.vogel.com.cn/2011/20100705/2142401.jpg[/img][b]结论[/b]经过柱净化的柴油和16种多环芳烃标样谱图如左图所示:红色谱图是柴油分析谱图,黑色谱图是16种多环芳烃标样谱图。从谱图可以看出,柴油在经过柱净化并浓缩后,其中多环芳烃能很好的实现检测。表1为16种多环芳烃的特征碎片离子
[B][size=4]前段时间我用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做塑料中的多环芳烃,用的柱子是hp-5ms,刚开始做的头几天峰形很好的,后来就逐渐开始拖尾,越来越拖得厉害,大约1000针样品后就彻底不行了, 但我再把它拿来做农残又仍然跟新柱子一样。衬管,柱子都是天天换割的。 只要再做多环芳烃就不行,不知道原因,修改的各种条件还是不行 后来我就买了个新ht-8的柱子,装上后做多环芳烃,立马就好了,但也是没过多久就两个月,就也拖尾得一塌糊涂。 再后来我又买了个hp-5msui的,现在正在用,也是有和上两个柱子一样的现象,才10天不到,现在已经开始拖尾了 不知道是否有人碰到和我一样的现象。可否有解决问题的方法?[/size][/B]
我最近在做关于某种细菌对多环芳烃降解率的摇瓶实验,只用了菲、芴、芘、荧蒽四种多环芳烃,在测溶液中多环芳烃残留量的时候,还需要加替代和内标吗?加的种类和测16种多环芳烃时一样吗?我们课题组以前都是做土壤重金属的,师兄师姐也不知道,文献里也没有说明白,很迷茫测16种多环芳烃时我用的替代是2-氟联苯和对三联苯混合液,内标是苊、菲、屈、苝的氘代物
我想测定石油组分中多环芳烃的含量。一般是经过正庚烷展开、烘干;再用甲苯展开、烘干;这样就有饱和份、芳香份、胶质和沥青质三个族组成。如果将多环芳烃和单环芳烃分离的话,是不是经过正庚烷、甲苯的展开,再用二甲基亚砜展开?关于用极性不同的溶剂展开,这句话在这个分析中是否这样理解?
液体石蜡中芳烃含量测定法(紫外分光光度法)此法中要求异辛烷以水为参比,在360-230处吸光度在0.00以下.但在实际使用中很多达不到这个要求,不知到用那个厂家的比较好,有没有提纯的办法?希望大家指点!
用多环芳烃的混标做标准曲线,其中16重组分分离度良好(未添加内标和替代物)。屈和苯并a蒽在285度左右出峰。用半挥发性有机物混标几十种的混标外加内标和替代物,进样。最后有两个多环芳烃,屈和苯并a蒽在245度左右出峰,但是降低出峰温度两种物质还是分不开。想请问还能怎么调整分析方式,使混标中这两个物质分开。。。
CNAS T0748橡胶制品中多芳烃环的中蒽含量测定不知道大家测试的结果是多少?可以讨论讨论
谁知道用气相色谱分析烷烃(馏程80-250度)组分中芳烃含量
多环芳烃污染物近年来受到人们的高度重视。美国EPA(国家环保总署) 确定了16种多环芳烃为代表检测物,并且推荐了检测方法。中国国标也给出了水中6种多环芳烃的检测方法。分别用乙腈/水或者甲醇/水梯度洗脱,都是用普通ODS柱。市场上很多品牌都有PAH检测专用柱,普通ODS柱检测问题在哪里呢?PAH检测专用柱有何优势呢?PAH专用柱特别的原因为何(比如是否可能是ODS填料键合一些芳基,瞎猜啊)?看了一些谱图,发现自来水或者土壤的PAH检测基线干扰挺严重的。或许是因为含量太少,萃取浓缩的原因吧。似乎针对水质土壤监控,有效的前处理方法更有意义。
日前欧盟委员会在官方公报中修订了食品中多环芳烃污染物以及重金属污染物有关的法规。 鉴于近期的科研数据以及欧盟食品安全局食物链污染物专家组在2008年采纳的意见,欧委会决定以食物中四种多环芳烃污染物的总含量作为评价多环芳烃污染的一个指标,这四种多环芳烃分别为:苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽、屈,另一个评价指标为苯并(a)芘的含量,以确保之前数据与以后数据的可比性。 与此同时,欧委会还在联合公报中修订了食品中苯并(a)芘以及重金属污染物的取样分析方法。 此次修订的生效日期为2012年9月1日。







 我要推广仪器
我要推广仪器
 下载APP
下载APP