前一阵子一直在做双酶切质粒重组,失败了 很多次,不过很快改善了 实验方法,用2周重组了 14个质粒。现就自己的体会,结合丁香园战友的宝贵经验,谈一下质粒重组的一些个人经验。1. 回收PCR产物 在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的 选择非常重要,尽量选择粘端酶切和 那些酶切效率高的限制酶,如BamHI、HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照:http://img.dxy.cn/upload/2006/08/13/31219184.pdf。双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒。酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/微升,在除外酶降解的 因素外,该酶可分解15 μg的DNA,而一般从1-4 ml菌液提出的 DNA约为3 μg,而PCR纯化后的产物(50体系)约为3 μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。2. 酶切、回收后的PCR产物与载体的连接摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03 pmol,后者取0.3 pmol。pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1 pmol 1000 bp DNA=0.66 μg,如载体是5380 bp,则0.03 pmol为0.03×5.38×0.66=0.106524 μg。测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0,另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的 MARKER每个条带约50 ng。连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。3. 转化a. 全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。b. 42℃加热45秒钟后,再在冰中放置1分钟。c. 加入890 μl AMP阴性培养基,37℃振荡培养60分钟。取100 μl铺板。也可离心后余100 μl。几个非常重要的问题1. 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解,为保险起见,一般连接3小时,16度。2. 对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干。3. 对照的设立:为验证双酶切是否成功,可做如下对照:A 酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性,如两管均成线性可初步判断双酶切成功。做转化时也要进行对照。设4个:A. 即拿双酶切的质粒产物也进行连接反应,这个对照可进一步看双酶切是否成功,如果长出克隆,说明很有可能只进行了单酶切,如没长出克隆,则证明双酶切成功,当然要保证感受态,培养基、连接酶都'正常'的情况下。B. 酶切过的未进行连接反应的双酶切产物,进行转化,这一步可以证明是否有残留的未被任何酶切的原始质粒。C. 设原始质粒为对照,意为检测整个操作过程中是否有误。D.AMP阴性板上用同一批感受态细胞铺板20微升足够,检测感受态状况。4. 所有的试剂切记低温保存 一步一个脚印,不要偷懒,图省事最后却更费事,注意设立对照。经PCR鉴定,克隆90%-100%的阳性率,所以在后面的 挑克隆中,我只挑选4个就足够了。然后双酶切鉴定,测序。
前一阵子一直在做双酶切质粒重组,失败了 很多次,不过很快改善了 实验方法,用2周重组了 14个质粒。现就自己的体会,结合战友的宝贵经验,谈一下质粒重组的一些个人经验。1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的 选择非常重要,尽量选择粘端酶切和 那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/微升,在除外酶降解的 因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的 DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。2、酶切、回收后的PCR产物与载体的连接摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03pmol,后者取0.3pmol。pmol为单位的DNA转换为为µg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为µg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为0.03×5.38×0.66=0.106524µg。测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的 MARKER每个条带约50ng。连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。3、转化:a、全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。 b、42℃加热45秒钟后,再在冰中放置1分钟。 c、加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl
第一节 概 述一. DNA的限制性内切酶酶切分析限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。它可分为三类:Ⅰ类和Ⅲ类酶在同一蛋白质分子中兼有切割和修饰(甲基化)作用且依赖于ATP的存在。Ⅰ类酶结合于识别位点并随机的切割识别位点不远处的DNA,而Ⅲ类酶在识别位点上切割DNA分子,然后从底物上解离。Ⅱ类由两种酶组成: 一种为限制性内切核酸酶(限制酶),它切割某一特异的核苷酸序列; 另一种为独立的甲基化酶,它修饰同一识别序列。Ⅱ类中的限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA的基础。绝大多数Ⅱ类限制酶识别长度为4至6个核苷酸的回文对称特异核苷酸序列(如EcoRⅠ识别六个核苷酸序列:5'- G↓AATTC-3'),有少数酶识别更长的序列或简并序列。Ⅱ类酶切割位点在识别序列中,有的在对称轴处切割,产生平末端的DNA片段(如SmaⅠ:5'-CCC↓GGG-3');有的切割位点在对称轴一侧,产生带有单链突出末端的DNA片段称粘性未端, 如EcoRⅠ切割识别序列后产生两个互补的粘性末端。5'…G↓AATTC…3' →5'… G AATTC…3'3'…CTTAA↑G …5' →3'… CTTAA G…5'DNA纯度、缓冲液、温度条件及限制性内切酶本身都会影响限制性内切酶的活性。大部分限制性内切酶不受RNA或单链DNA的影响。当微量的污染物进入限制性内切酶贮存液中时,会影响其进一步使用,因此在吸取限制性内切酶时,每次都要用新的吸管头。如果采用两种限制性内切酶,必须要注意分别提供各自的最适盐浓度。若两者可用同一缓冲液,则可同时水解。若需要不同的盐浓度,则低盐浓度的限制性内切酶必须首先使用,随后调节盐浓度,再用高盐浓度的限制性内切酶水解。也可在第一个酶切反应完成后,用等体积酚/氯仿抽提,加0.1倍体积3mol/L NaAc和2倍体积无水乙醇,混匀后置-70℃低温冰箱30分钟,离心、干燥并重新溶于缓冲液后进行第二个酶切反应。DNA限制性内切酶酶切图谱又称DNA的物理图谱,它由一系列位置确定的多种限制性内切酶酶切位点组成,以直线或环状图式表示。在DNA序列分析、基因组的功能图谱绘制、DNA的无性繁殖、基因文库的构建等工作中,建立限制性内切酶图谱都是不可缺少的环节,近年来发展起来的RFLP(限制性片段长度多态性)技术更是建立在它的基础上。构建DNA限制性内切酶图谱有许多方法。通常结合使用多种限制性内切酶,通过综合分析多种酶单切及不同组合的多种酶同时切所得到的限制性片段大小来确定各种酶的酶切位点及其相对位置。酶切图谱的使用价值依赖于它的准确性和精确程度。在酶切图谱制作过程中,为了获得条带清晰的电泳图谱,一般DNA用量约为0.5-1μg。限制性内切酶的酶解反应最适条件各不相同,各种酶有其相应的酶切缓冲液和最适反应温度(大多数为37℃)。对质粒DNA酶切反应而言, 限制性内切酶用量可按标准体系1μg DNA加1单位酶,消化1-2小时。但要完全酶解则必须增加酶的用量,一般增加2-3倍,甚至更多,反应时间也要适当延长。二. 凝胶电泳琼脂糖或聚丙烯酰胺凝胶电泳是分离鉴定和纯化DNA片段的标准方法。该技术操作简便快速,可以分辨用其它方法(如密度梯度离心法)所无法分离的DNA片段。当用低浓度的荧光嵌入染料溴化乙啶(Ethidium bromide, EB)染色,在紫外光下至少可以检出1-10ng的DNA条带,从而可以确定DNA片段在凝胶中的位置。此外,还可以从电泳后的凝胶中回收特定的DNA条带,用于以后的克隆操作。琼脂糖和聚丙烯酰胺可以制成各种形状、大小和孔隙度。琼脂糖凝胶分离DNA片度大小范围较广,不同浓度琼脂糖凝胶可分离长度从200bp至近50kb的DNA片段。琼脂糖通常用水平装置在强度和方向恒定的电场下电泳。聚丙烯酰胺分离小片段DNA(5-500bp)效果较好,其分辩力极高,甚至相差1bp的DNA片段就能分开。聚丙烯酰胺凝胶电泳很快,可容纳相对大量的DNA,但制备和操作比琼脂糖凝胶困难。聚丙烯酰胺凝胶采用垂直装置进行电泳。目前,一般实验室多用琼脂糖水平平板凝胶电泳装置进行DNA电泳。琼脂糖主要在DNA制备电泳中作为一种固体支持基质,其密度取决于琼脂糖的浓度。在电场中,在中性pH值下带负电荷的DNA向阳极迁移,其迁移速率由下列多种因素决定:1、DNA的分子大小:线状双链DNA分子在一定浓度琼脂糖凝胶中的迁移速率与DNA分子量对数成反比,分子越大则所受阻力越大,也越难于在凝胶孔隙中蠕行,因而迁移得越慢。2、琼脂糖浓度一个给定大小的线状DNA分子,其迁移速度在不同浓度的琼脂糖凝胶中各不相同。DNA电泳迁移率的对数与凝胶浓度成线性关系。凝胶浓度的选择取决于DNA分子的大小。分离小于0.5kb的DNA片段所需胶浓度是1.2-1.5%,分离大于10kb的DNA分子所需胶浓度为0.3-0.7%, DNA片段大小间于两者之间则所需胶浓度为0.8-1.0%。
喷了几次了,总是出现筛子孔。孔经常先在样品边缘出现,然后才在中间出现,并且需要等待几十秒才能透光。。。这是怎么回事,求指点!还有就是束流用大的还是用小的?样品时变形态高纯镁(沿挤压方向压缩的纯镁挤压棒),圆片厚度50-60微米双喷液是5.3g氯化锂+11.16g高氯酸镁+100mL乙二醇丁醚+500mL甲醇双喷温度-55℃~-30℃(这个温度段的都试过),电压100v,电流50毫安左右
酶切反应建议一个版友提供的简述版的酶切反应建议,还是很不错的,很适合初步做酶切反应者。一、 建立一个标准的酶切反应二、 选择正确的酶三、 酶 四、 DNA 五、 缓冲液六、 反应体积七、 混合 八、 反应温度九、 反应时间 十、 终止反应 十一、贮存 十二、稳定性 十三、对照反应 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=158046]酶切反应建议[/url]
请教下液相检测胰蛋白酶酶切效果,检测条件和柱子类型?
[color=#444444]采用液相色谱仪检测,使用氨水或0.01%的庚烷磺酸钠作为流动相都检测不到霜霉威的峰,请问哪位大神检测过霜霉威?我是做分析检测的,急切寻找方法[/color]
请教大家一个问题,我问同学要了个1301的质粒,用BglII 和EcoRI切出4条带来,本来应该两条带。而且我可以保证酶切是完全的。可同学说他给我的质粒没问题,前几天他用还是好的。高手给解决一下吧!!
小弟在用安捷伦6460C做 SN/T 0525-2012 这个方法 现在遇到这个问题 我把福美双标准物质配置成10ug/mL的标准溶液后,进行质谱全扫 之后提取241.2 这个母离子发现是啥都没有。。 小弟现在不知道该咋做了 在此向诸位大神请求做福美双的这个方法。 附件中是我做的全扫图
请求各位大大,本人是酶切的新手,有问题想请教一下关于基因分型的,我以前没学过做相关的实验,只是看文献去进行实验。CDH23-rs 3802711 Upper 5’- CCA CAG TTC CTG CCA ATG -3’ Lower-5’-GTC CAG CTC CAC GTC CTT -3’该基因PCR后的目的片段大小是115bp; PCR后用限制性内切酶MSP 1 进行酶切后,出现多大的条带分成多少段是 TT型, CC型,CT型; CDH23-EXON7 Upper 5’- TCT TCA TCA ACC TGC CTT AC-3’ Lower-5’-GCT GTG CCA GAA CAC TCA T-3’该基因PCR后的目的片段大小是202bp; PCR后用限制性内切酶MSP 1 进行酶切后,出现多大的条带或者分成多少段是 AA型 GG型 AG型;谢谢各位大大
有哪位老师用液质做过福美双的?母离子、子离子是多少呀?分子量+1的母离子峰241.5找不到呢?
请教高手,用液相做福美双的检测,我是用乙腈提取,过4g弗罗里硅土柱子,结果都没回收,有人做过吗!柱子应该没有问题,大家帮忙分析一下吧!!万分感谢
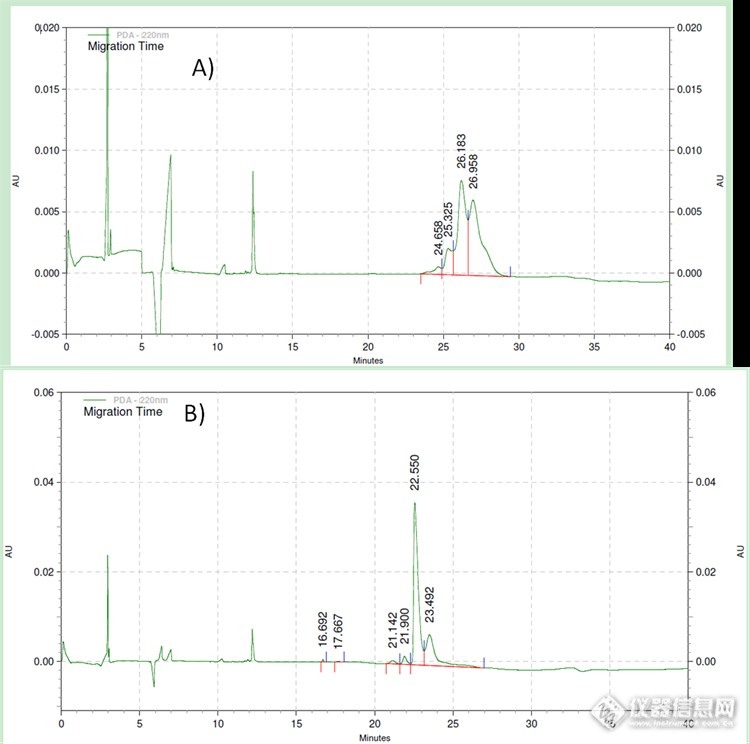
双抗如何跑CE?跑出来的如图A轮廓。(不要关心保留时间)融合蛋白我们是用PNGase F酶切,切下后的峰(有四个峰,图B)分别是什么呢?http://ng1.17img.cn/bbsfiles/images/2016/11/201611211119_01_3115342_3.png
想请教一下有人用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做过福美双和硫醇吗?福美双我查了一下物理性质,只有熔点,没有沸点.是不是沸点很高,或者不稳定啊?而且我看蔬菜,水果中检测福美双的国标是用分光光度法,不知道是不是因为[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]不能做.还有有高手做过硫醇吗?不知道会不会很臭啊?希望有高手能予以指教,非常感谢.
大家有没有做过福美双的检测,我用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS走样,标液都可以走出来,但是经过前处理后,都没回收,我是用GPC净化的。前处理过程是样品乙腈提取,旋转蒸干,乙酸乙酯:环己烷复溶,GPC净化,旋转蒸发,丙酮定容上机。大家能帮忙分析是什么原因吗?祈求有相关经验不吝赐教。
请问,我在做蛋白质谱的过程中,酶切膜蛋白时,总有一些序列cover不到(哪怕胶内酶解PSM很高时,也没有改善)。请教有没有大佬传授一些经验,谢谢
有谁知道2水双乙酰丙酮镁的性质,或者是其相关资料?谢谢了!
请问有谁知道双乙酰丙酮镁的分析方法?谢谢了!
请问谁有以下标准可否给我一份,谢谢了甲基硫菌灵的标准HG 2462.2-1993福美双的标准HG3758-2004
2012年1月6日加拿大发布通报,加拿大卫生部有害生物管理局(PMRA)拟定霜霉威盐酸盐(Propamocarb hydrochloride)国内最大残留限量。 MRL (ppm) 农原料品(RAC)及/或加工品 200 散叶莴苣 150 结球莴苣 5.0 番茄酱 2.0 番茄* *因田番茄纳入TATOO真菌标签内容,建议替代目前番茄规定的0.01ppm的MRL。
切了柱子,出峰时间会提前。 想保证保留时间与没切割前一致怎么办呢, 降低初始柱温好不好?
霜霉威出双峰?流动相,方法都没变
有那位高人做过福美双残留量的气相色谱法测定,请指教
用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]在代森锰锌和福美双的标样中分别扫到了159和140的前提离子,配制的是单标,响应不错,不清楚是不是目标物,求助!
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=125790]防霉防腐剂双乙酸钠的合成研究[/url]希望对部分人有用!
请教一下,谁有福美双可湿性粉剂在小麦上的安全间隔期,谢谢!
问题: 有谁知道PNgaseF酶切糖后的终止是用什么条件比较合适?
Eppendorf的pcr仪器用了好几年了,最近出现0error,只是恒温做酶切有问题吗?
霜霉威检测方法1.分析目标化合物霜霉威、霜霉威盐酸盐2.仪器设备带碱热离子检测器或高灵敏度氮磷检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]—质谱仪。3.试剂使用附录2所列试剂。4.标准品霜霉威:含霜霉威99%以上,熔点为45℃~55℃。5.试验溶液的制备谷类:将样品粉碎,通过420μm的标准网筛后,称取其20.0g,加入30mL 1mol/L盐酸,放置2小时。水果和蔬菜:准确称取约1 kg样品,必要时定量加入适量水,搅碎混合均匀后,称取相当于20.0g样品的量。加入80 mL丙酮:水(7:3)混合溶液(谷类为50mL),搅拌5分钟后,用涂布1cm厚硅藻土的滤纸抽滤于磨口减压浓缩器中。取出滤纸上的残留物,加入50mL丙酮:水(7:3)混合溶液,搅拌5分钟后,按上述同样操作,合并滤液于减压浓缩器中,40℃以下浓缩至约40mL。将其移入预先注入50mL乙醚和5g氯化钠的200mL分液漏斗中,振摇混合1分钟后,静置,弃去乙醚层。水层中加入50mL乙醚,按上述同样操作,水层移入200mL分液漏斗中。水层中加入5g无水碳酸钠(谷类为8g)和50mL乙醚,用振荡器激烈振荡5分钟后,静置,乙醚层移入200mL三角瓶中。水层中加入50mL乙醚,按上述同样操作,重复2次,合并乙醚层于上述三角瓶中。加入适量无水硫酸钠,不时振摇、混合,放置15分钟后,滤入磨口减压浓缩器中。再用10ml乙醚洗涤三角瓶,以此洗液洗涤滤纸上的残留物,重复操作两次。合并两洗液于减压浓缩器中,40℃以下浓缩至约1mL,室温下通空气进一步干燥。残留物中加入乙酸乙酯溶解,准确至1mL,此为试验溶液。6.操作方法a 定性试验按下列操作条件进行试验,试验结果应与标准品的一致。操作条件柱:内径0.25mm、长30m的石英毛细管,涂布0.25μm厚[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]用14%氰丙基苯基—甲基硅酮,老化。柱温:在60℃保持1分钟,此后每分钟升温10℃。到达160℃后保持2分钟,再每分钟升温4℃。到达180℃后每分钟升温20℃,到达260℃后保持5分钟。进样器温度:250℃进样方式:不分流检测器温度:250℃气体流量:以氦气作载气。调节流速使霜霉威约14分钟30秒流出。调节空气和氢气的流量至适当条件。b 定量试验根据与a 定性试验相同试验条件所得的试验结果,峰高法或峰面积法定量。c 确证试验按照与a 定性试验相同的试验条件,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]—质谱仪测定,试验结果应与标准品的一致。此外,必要时用峰高法或峰面积法进行定量。7.定量限0.01 mg/kg8.注意事项霜霉威的分析值包括霜霉威和霜霉威盐酸盐。9.参考文献无
有块仕富梅 5400双检测器的 ,一个是氧化锆,一个是磁氧,他们的检测通道是否可以互换?有知道的告诉一声 我想做个试验的。谢谢啦。







 我要推广仪器
我要推广仪器
 下载APP
下载APP