“植物奶油危机事件”引发了消费者的恐慌。中国焙烤食品糖制品工业协会理事长朱念琳表示,消费者不必谈“氢化植物油”色变,现在消费者存在两个误区:一是“氢化植物油”即植物奶油、起酥油等,并不能和反式脂肪酸完全画等号,经过工艺改进的氢化植物油可以降低反式脂肪酸,甚至不含反式脂肪酸;二是应该限制食品中“反式脂肪酸”的含量,但并不是要禁止使用氢化植物油,在美国、丹麦等也未出台限制使用氢化植物油,仅是建议居民日均反式脂肪酸摄入量不超过总能量的1%。 朱念琳介绍,以制作糕点为例,酥皮、起酥等均需要酥油来起酥,过去人们通过熬制猪油、牛油等动物性油脂来烹制。由于担心存在于荤油中的胆固醇可能会对心脏带来威胁,上世纪80年代,氢化植物油开始在中国食品加工业使用,植物油因高温不稳定及无法保存等问题,从国外引进了氢化技术,有研究表明高温脱臭后的油脂中反式脂肪酸的含量可增加1%至4%。 世界卫生组织和联合国粮农组织在《膳食、营养与慢性病》中建议,以每天摄取2000千卡热量的人为例,每日摄取的总脂肪应为66克,饱和脂肪酸22.2克,反式脂肪酸2.2克以下。 不同品牌氢化植物油的质量优劣区别很明显,朱念琳说,氢化植物油加工工艺需要改进,食品加工企业的制作也要研发新的方式,但是这需要一个过程。 现在市面上有氢化大豆油、氢化棕榈油、氢化椰子油……目前可以通过酶法或化学催化剂、配方调整、改进氢化工艺等方式在一定程度上减低反式脂肪酸的含量。如果植物油完全氢化,完成从液态变为固态,就可以做到反式脂肪酸含量几乎为零。问题在于完全氢化的植物油制作植物奶油、起酥油等,厚厚的一层根本无法涂抹。他希望这次的“植物奶油危机事件”能成为一个巨大的推动力,尽快帮助食品加工企业加强科研经费的投入,生产出更安全、美味的食品。 以往“动物奶油”意味着饱和脂肪含量高,不健康等,如今却成为商家吸引眼球的卖点。记者在一家蛋糕网站上看到,一行“不含植物氢化油”的字正在闪烁。网站称所有蛋糕均采用“动物奶油”。 中国焙烤食品糖制品工业协会呼吁,希望卫生部等有关部门尽快出台限制反式脂肪酸的相关标准,让企业生产中有法可依。也呼吁食品企业加强科研经费投入,研究食品生产过程中控制反式脂肪酸产生的技术,推出企业使用反式脂肪酸含量低或不含反式脂肪酸的食用油脂,降低食品中反式脂肪酸的含量。提倡食品企业标识营养标签,注明反式脂肪酸的含量,让消费者明明白白消费。
请教有没有氢化物-原子荧光法测水样中汞的方法。手头的都是用BR消解水样,用盐酸羟胺还原过量溴,再用氯 化亚锡还原二价汞的方法。没有氢化物发生法方便。不知有没有这类的标准可查
为什么用原吸氢化物测尿苷酸二酸中砷时样品加入硫脲后会有沉淀生成?
近日,“植物奶油危机事件”引发了消费者的恐慌。昨天,中国焙烤食品糖制品工业协会理事长朱念琳表示,消费者不必谈“氢化植物油”色变,现在消费者存在两个误区:一是“氢化植物油”即植物奶油、起酥油等,并不能和反式脂肪酸完全画等号,经过工艺改进的氢化植物油可以降低反式脂肪酸,甚至不含反式脂肪酸;二是应该限制食品中“反式脂肪酸”的含量,但并不是要禁止使用氢化植物油,在美国、丹麦等也未出台限制使用氢化植物油,仅是建议居民日均反式脂肪酸摄入量不超过总能量的1%。 ■上世纪80年代使用氢化植物油 朱念琳介绍,以制作糕点为例,酥皮、起酥等均需要酥油来起酥,过去人们通过熬制猪油、牛油等动物性油脂来烹制。由于担心存在于荤油中的胆固醇可能会对心脏带来威胁,上世纪80年代,氢化植物油开始在中国食品加工业使用,植物油因高温不稳定及无法保存等问题,从国外引进了氢化技术,有研究表明高温脱臭后的油脂中反式脂肪酸的含量可增加1%至4%。 世界卫生组织和联合国粮农组织在《膳食、营养与慢性病》中建议,以每天摄取2000千卡热量的人为例,每日摄取的总脂肪应为66克,饱和脂肪酸22.2克,反式脂肪酸2.2克以下。
有人做过2015版中国药典的氢化蓖麻油脂肪酸组成吗?为什么会12羟基硬脂酸甲酯偏低呢?前处理的回流萃取,哪一步需要注意呢?

http://ng1.17img.cn/bbsfiles/images/2010/11/201011081110_258052_1641058_3.jpg 氢化油,也被叫做“植物奶油”“植物黄油”“植脂末”。目前,在面包、奶酪、人造奶油、蛋糕和饼干等食品焙烤领域广泛使用。氢化油产生大量反式脂肪酸,增加心血管疾病、糖尿病等风险,世界各国已纷纷限制,但中国却在大规模、无限制地使用。(据11月7日央视) 在饼干、蛋糕、麦片、方便面等包装上,都印刷着含有氢化植物油,却没有标注上具体含量。也就是说,我每天都在吃含氢化油的食品,却不知道吃了多少。可见,要拆除健康炸弹的隐忧,关键之处是尽快制定食品标准,限制氢化油的含量。 氢化油与食品添加剂一样,属于额外的东西,对于食品本身来讲,并没有提高其营养价值,只是增加食品的美味可口,丰富了我们的味觉。这是食品工业化的伟绩,也是为迎合消费者的食欲,从而采用化学方法改变食品成分。科学本来就是把双刃剑,是益是害,要看怎么运用了。氢化油诞生百年来,由于其应用广泛,使食物变得更加松软酥脆,一直受到消费者的青睐,而发现其潜存的危害性,也是最近十来年的事。 氢化油对健康主要有四个方面的危害:增加血液黏稠度和凝聚力,促进血栓形成;提高低密度脂蛋白胆固醇,促进动脉硬化;增加糖尿病的发病率;影响婴幼儿和青少年正常的生长发育,并可能对中枢神经系统发育产生不良影响。氢化油会产生大量反式脂肪酸,据健康专家介绍,一般的脂肪吃在身体里7天就代谢了,而反式脂肪吃在身体里50天才可以代谢,这就是为什么洋快餐会导致肥胖的原因。 如同其它对健康有危害的东西一样,总是在经过一段时间后才被发现。氢化油的危害性,经过媒体的报道,应该会引起消费者的警觉心。但是,受到现代社会生活方式影响,消费者与食品加工生产之间有隔膜,根本不了解也弄不懂,食品里面到底含有哪些有害成分。这就需要依赖于食品监管部门,由它们为消费者把关,通过制定严格的食品标准,限制有害成分的含量。 食品安全危机的案例,我们已经遭遇过许多次,心理承受力亦被锻炼成世界一流水平。氢化油属于潜藏的健康炸弹,其危害性并不显眼,甚至因美味而俘获无数消费者。因此,对于拆除氢化油的危害性,除了制定含量标准外,还需采取多种辅助手段,包括普及健康知识、宣传健康饮食习惯、全程监管氢化油生产企业等等。这项健康工程,需要尽快启动,不能再拖延时日,以免造成更大的健康危机。12楼:《经济半小时》报道34楼:卫生部正评估植物奶油风险 婴幼儿食品禁用42楼:我国居民的反式脂肪酸人均摄入量在0.6克左右,远低于欧美国家的水平。
钨铁合金中砷、锡、铅、锑、铋的测定(氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法)一、 方法提要试样以草酸-双氧水分解,定容,作为母液。分取母液在不同的条件下利用氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法测定砷、锡、铅、锑、铋的含量。二、 仪器及工作条件2.1 WYX-402C型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计(沈阳分析仪器厂)2.2 WHG-102A2型流动注射氢化物发生器(北京瀚时制作所)2.3 高性能砷空心阴极灯2.4 高性能锑空心阴极灯2.5 普通铅空心阴极灯2.6 普通铋空心阴极灯2.7 普通锡空心阴极灯2.8 工作条件元素分析线nm灯电流Ma光谱通带nm载气流量ml/min石英管温℃As193.710~120.480~120900Sn286.38~100.4300900Sb217.610~120.480~100900Pb283.36~80.4150900Bi223.06~80.4100900三、 试剂3.1 草酸 3.2 过氧化氢3.3 盐酸3.4 醋酸(GR)3.5 抗坏血酸3.6 碘化钾3.7 硼氢化钾(1.5%)称取1.5g硼氢化钾,约0.3g氢氧化钠(稳定剂)倒入塑料瓶中,加水100ml溶解。室温下可用一周。3.9 载液:盐酸(1% V/V)3.10 醋酸钠3.11 载气:纯氮或纯氩。流量见“各元素特有条件”。3.12 砷标准溶液(1μg/ml):将0.1320g三氧化二砷用100ml盐酸溶解,用水稀至1000ml。密封保存。分取1.00ml于100ml容量瓶中,以10%的盐酸溶液稀至刻度,现用现配。3.13 锡标准溶液(2μg/ml):将0.1000高纯金属锡溶解于100ml浓盐酸中,然后用水稀至1000ml,密封保存。分取1.00ml于100ml容量瓶中,以PH3.7的醋酸-醋酸钠溶液稀至刻度,现用现配。3.14 锑标准溶液(2μg/ml):将0.1000g高纯金属锑用10ml硝酸和5ml盐酸溶解,待完全溶解后以水稀至1000ml。密封保存。分取2.00ml于100ml容量瓶中,以10%的盐酸溶液稀至刻度,现用现配。3.15 铋标准溶液(2μg/ml):将0.1000g高纯铋溶解于130ml硝酸(1+1)溶液中,溶解完全后,以水稀至1000ml,密封保存。分取2.00ml于100ml容量瓶中,以20%(V/V)盐酸溶液稀至刻度。现用现配。3.16 铅标准溶液(1μg/ml):将0.1000g高纯铅粉溶解于少量6mol/L硝酸中,以水稀至1000ml。分取1.00ml于100ml容量瓶中,以水稀至刻度。现用现配。四、 试样 应通过200目孔筛五、 分析步骤5.1 试样量 称取0.1000~0.2000g试样5.2 空白试验随同试样做空白试验5.3 测定 5.3.1将试样置于250ml烧杯中, 加入3~4g固体草酸,以15ml过氧化氢分次加入,加热低温分解完全,在有少量气泡时(不能出现钨酸沉淀,试液清亮即可。),取下,稍冷,移入50ml容量瓶中,以水稀至刻度。即为母液。5.3.2砷的测定:5.3.2.1分取母液0.5~2.0ml于50ml容量瓶中,加碘化钾浓度为0.5~1%,加入10%盐酸溶液,沸水浴中加热约10min(温度升到约80~90℃),冷却后,加抗坏血酸到浓度为0.2~0.5%后以10%的盐酸溶液定容,测定。5.3.2.2曲线的制作:分取标准溶液(1.0μg/ml)0.00/0.25/0.50/0.75/1.00/1.25ml于一组50ml容量瓶中,以下按4.3.2.1步骤操作。5.3.3锡的测定:5.3.3.1分取母液0.5~2.0ml于50ml容量瓶中,以PH3.7的醋酸-醋酸钠溶液定容,测定。5.3.3.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.3.1步骤操作。5.3.4锑的测定:5.3.4.1分取母液0.5~2.0ml于50ml容量瓶中,加碘化钾浓度为0.5~1%,加入10%盐酸溶液,放置1~2min,加抗坏血酸到浓度为0.2~0.5%后以10%的盐酸溶液定容,测定。5.3.4.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.4.1步骤操作。5.3.5铋的测定:5.3.5.1分取母液0.5~2.0ml于50ml容量瓶中,加入醋酸溶液1.0ml,以20%(V/V) 的盐酸定容。5.3.5.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.5.1步骤操作。5.3.6铅的测定:5.3.6.1分取母液0.5~2.0ml于50ml容量瓶中,加入过氧化氢溶液0.5~1.0ml,醋酸溶液1.0~2.0ml,以水定容。5.3.6.2分取标准溶液(1.0μg/ml)0.00/0.25/0.50/0.75/1.00/1.25ml于一组50ml容量瓶中,以下按4.3.6.1步骤操作。六、 分析结果的计算:按下式计算砷锡锑铅铋的百分含量: As、Sn、Sb、Pb、Bi(%)=m1/m0r ×100 试中 m1------------从工作曲线上查出的砷锡锑铅铋量(g) m0------------试样量(g) r---------试液分取比
氢化物消除干扰的途径 在干扰的类型及机理确定以后,即可采取相应的措施来消除和降低干扰,消除分析中常见的干扰,可以通过以下途径。 (一)选择最佳酸度介质 因为氢化物的反应是在酸性介质中进行,因此有关研究报千都要涉及反应液介质问题,通常采用盐酸介质,其浓度对砷、锑、铋的影响不大,允许浓度范围较宽。但随酸的性质不同而有不同的影响,如硝酸对砷和硒的测定有干扰,酸的浓度对硒和碲的测定有影响,要有较高的酸度,而锗、锡、铅的氢化物发生都要求在较严格折浓度下进行测定。 表一 氢化物反应酸度条件 元素 价态 反应介质 元素 价态 反应介质As +3 1-6NHCl Sb +3 1-6NHCl Te +4 4-6NHCl Bi +3 1-6NHCl Ge +4 20%H3PO4 Se +2 1-6NHCl Sn +4 酒石酸缓冲溶液PH=1.3 控制酸度也可以控制价态干扰,例如在PH=4个时,只有三价砷能转化为砷化氢,而当PH=5时,三价砷和五价砷均可还原成砷化氢,则可测出两种价态砷的总量。 (二)选择最佳还原 剂及用量 有人对比采用Zn和硼氢化钠两种还原体系时,不同离子的干扰效应,发现某些离子的干扰程度取决于所采用的还原剂种类。他们还研究了测定砷和锑时碘化钾对共存离子的掩敝作用,掩敝效果与硼氢化钠的浓度有极密切的关系,总的来说,低的硼氢化钠溶液可以降低共存离子的干扰,原因在于稀溶液不能将金属离子还原成金属。需要指出的是,硼氢化钠溶液不能消除其它可形成氢化物元素的干扰。 (三)利用掩敝作用 对于重金属及贵金属的干扰,除通过选择最佳的酸介质和还原剂用量外,还可以加入适当的络合剂,利用对共存离子的掩敝作用,防止共存离子与待测元素生成难溶的化合物或避免被硼氢化钠还原成沉淀析出,因而可提高氢化物的释放效率。 表二 测定不同元素加入的掩蔽剂 测定元素 干扰元素 加入试剂As Se Cu、Co、Ni、Fe EDTA Bi Ni EDTA As Ni KCNS Te Cu、Au 硫脲Bi Cu 硫脲Bi Cu KI As Ni 1、10邻菲罗琳、氨基硫脲As Cu、Co、Ni等 8-羟基奎琳 As Cu K4[Fe(CN)6] Sn Cu、Ni、Fe 硫脲-抗坏血酸 另外,仍有大量的掩敝剂已广泛用来克服氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中的干扰,可以参考有关资料。 (四)共沉淀和浮选分离 共存离子的干扰可以通过共沉淀和浮选分离等预处理等方法加以克服,如测定地下水中的砷,海水中的锡、砷、铋,河水中的砷、铋、自来水的锑,都可以用Fe(OH3共沉淀和浮选分离的公方克服共存离子的干扰,测定金属铜中的铋、砷、硒、锡、碲可以借助氢氧化镧共沉淀来克服铜的干扰,测定饮用水中的Pb可以通过二氧化锰共沉淀克服铜和镍的干扰。也可以用锡的测定。 (五)电解和溶剂萃取分离 测定金属中杂质元素往往需要预分离,电解方法也是一种良好的分离方法,但由于操作繁琐而不常用,溶剂萃取分离是一种有效的分离方法,现已广泛用于氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]方法,萃取是一项十分丰富的学科,所报道的文献也较多,可以参考其它的资料。 (六)色谱分离 为了进一步消除基体成分的干扰和进行金属化学形态的分析,目前发展有色谱-[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]联用技术,这种技术综合了色谱分离效果好的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]检测灵敏度高的特点。由于许多实验室不具有色谱仪,仪器联接有一定的技术。目前,这种分析方法并没有得到广泛的发展。 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析的干扰是多种多样的。充分认识干扰的机理,采用有效的清除干扰的方法是近代[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分析中的关键问题,特别是环境、水样、食品等痕量元素的分析中。
有哪位大神做过2015版中国药典的氢化蓖麻油脂肪酸组成呢?为什么我做出来俩结果不平行 两针对比下来有一针12羟基硬脂酸甲酯偏低 而硬脂酸甲酯偏高 难道二者之间存在转化关系么?还有实验过程中有什么需要注意的吗?
公司使用氢化物发生器+[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]进行Hg和As的测定,以下是本人的若干使用经验。现在珠江三角洲附近使用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]+氢化物发生器来测定八大重金属的小工厂越来越多(暂且不论检出限等),但是在测定Sb,Se,Hg和As基本上都有些小问题。以下抛砖引玉。测定的样品主要为微波消解的皮革,织物或电热板消解的涂层,油墨。一:关于样品叙述:1.涂层主要是油漆样品,油漆主要为化工原料(邻苯二甲酸酯等),填料等构成,在电热板等比较温和的条件下消解不完全,因此样品粘度较大,进样品时较容易产生泡沫。只能采用稀释或者添加酸,消泡剂等加以改善。2.皮革和织物样品绝大部分或大部分为天然纤维或蛋白质,在微波助酸消解下,样品一般为澄清,消解完全。3.油墨样品, 油墨连接剂也是油墨的主要成分之一,起分散色料和辅助料的媒介作用,是由少量天然树脂、合成树脂、纤维素、橡胶衍生物等溶于干性油或溶剂中制得。有一定的流动性,使油墨在印刷后形成均匀的薄层,干燥后形成有一定强度的膜层,并对颜料起保护作用,使其难以脱落.因此油墨样品也较易产生泡沫,缓解措施同上。二:关于氢化物发生器国内氢化物发生器配套厂家主要有北京翰时,上海光谱,北京普析等。国内厂家出于节省成本的考虑,发生器较为简单,其机器主要构成是减压阀,气液分离器,玻璃转子流量计,反应线圈等部件。国外厂家如varian原厂发生器主要还包含有蠕动泵,可更换的反应线圈。国内厂家的机器普遍存在稳定性较差,管路不能进行更换,易老化,进样效率低等缺点。三:分析技巧1.标准溶液配置:As标准溶液必须为三价砷,五价同样条件下测试吸光度约只有三价标准溶液的1/5左右,国内纳克等公司标准溶液一般为As2O3与NaOH熔融后,用盐酸定容,可以满足测试要求。国外MECRK如为特殊说明一般为五价砷,我们实验室两种标液均有,所以有时不同人配置的标液会截然不同。日本WOKA标液也为三价。2.配置Hg和As标准溶液,可以采用多级稀释配置。如我们配置1ug/l,2ug/l,5ug/l的标准溶液时,可以采用先从1000mg/l的标准溶液配置成1000ug/l的母液,再从1000ug/l配置要求所需的标准溶液。配置时尽量采用同一支[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url],容量瓶用较大规格如100ml。5ug/lHg的吸光度(硝酸基体)可达0.1左右。3.测定汗液基体样品时,标准曲线应该添加少量硝酸(约1%即可)以增加酸度,提高稳定性。4.测试时间设置:由于样品需要进入反应线圈充分混合,经气液分离后进入洗吸收池,所以进样时间应尽量设置较长。我们设置时间为80s,消耗样品约为9ml,重复性,精密度等均较好。5.关于价态还原:用5%硫脲-5%抗坏血酸还原As(Ⅴ)、Sb(Ⅴ) 为 As(Ⅲ)、Sb(Ⅲ),控制酸度为10%HCl测定As和Sb。测定Se 应单独称样,用混酸溶解 ,在1+1HCl中于水浴上加热半小时还原Se(Ⅵ)至Se(Ⅳ)。6.关于还原剂:一般使用的还原剂主要有硼氢化钠,硼氢化钾及稳定剂氢氧化钠。典型的还原剂配置为硼氢化钠1%~2.5%,氢氧化钠0.5%。通常认为硼氢化钾活性较硼氢化钠高。7.关于稀硝酸(10%)对As(Ⅲ)的氧化性。本人分别使用纳克,WOKA三价标液,稀硝酸(10%)做为基体配置1PPM的砷标液,在室温下静置10昼夜,配置为5PPB的浓度后进行测试,吸光度没有发现有明显变化。所以稀硝酸在室温下几乎不对三价砷标液有氧化作用。
钨铁合金中砷、锡、铅、锑、铋的测定(氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法)一、 方法提要试样以草酸-双氧水分解,定容,作为母液。分取母液在不同的条件下利用氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法测定砷、锡、铅、锑、铋的含量。二、 仪器及工作条件2.1 WYX-402C型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计(沈阳分析仪器厂)2.2 WHG-102A2型流动注射氢化物发生器(北京瀚时制作所)2.3 高性能砷空心阴极灯2.4 高性能锑空心阴极灯2.5 普通铅空心阴极灯2.6 普通铋空心阴极灯2.7 普通锡空心阴极灯2.8 工作条件元素 分析线nm 灯电流Ma 光谱通带nm 载气流量ml/min 石英管温℃As 193.7 10~12 0.4 80~120 900Sn 286.3 8~10 0.4 300 900Sb 217.6 10~12 0.4 80~100 900Pb 283.3 6~8 0.4 150 900Bi 223.0 6~8 0.4 100 900 三、 试剂3.1 草酸 3.2 过氧化氢3.3 盐酸3.4 醋酸(GR)3.5 抗坏血酸3.6 碘化钾3.7 硼氢化钾(1.5%)称取1.5g硼氢化钾,约0.3g氢氧化钠(稳定剂)倒入塑料瓶中,加水100ml溶解。室温下可用一周。3.9 载液:盐酸(1% V/V)3.10 醋酸钠3.11 载气:纯氮或纯氩。流量见“各元素特有条件”。3.12 砷标准溶液(1μg/ml):将0.1320g三氧化二砷用100ml盐酸溶解,用水稀至1000ml。密封保存。分取1.00ml于100ml容量瓶中,以10%的盐酸溶液稀至刻度,现用现配。3.13 锡标准溶液(2μg/ml):将0.1000高纯金属锡溶解于100ml浓盐酸中,然后用水稀至1000ml,密封保存。分取1.00ml于100ml容量瓶中,以PH3.7的醋酸-醋酸钠溶液稀至刻度,现用现配。3.14 锑标准溶液(2μg/ml):将0.1000g高纯金属锑用10ml硝酸和5ml盐酸溶解,待完全溶解后以水稀至1000ml。密封保存。分取2.00ml于100ml容量瓶中,以10%的盐酸溶液稀至刻度,现用现配。3.15 铋标准溶液(2μg/ml):将0.1000g高纯铋溶解于130ml硝酸(1+1)溶液中,溶解完全后,以水稀至1000ml,密封保存。分取2.00ml于100ml容量瓶中,以20%(V/V)盐酸溶液稀至刻度。现用现配。3.16 铅标准溶液(1μg/ml):将0.1000g高纯铅粉溶解于少量6mol/L硝酸中,以水稀至1000ml。分取1.00ml于100ml容量瓶中,以水稀至刻度。现用现配。四、 试样应通过200目孔筛五、 分析步骤5.1 试样量称取0.1000~0.2000g试样5.2 空白试验随同试样做空白试验5.3 测定 5.3.1将试样置于250ml烧杯中, 加入3~4g固体草酸,以15ml过氧化氢分次加入,加热低温分解完全,在有少量气泡时(不能出现钨酸沉淀,试液清亮即可。),取下,稍冷,移入50ml容量瓶中,以水稀至刻度。即为母液。5.3.2砷的测定:5.3.2.1分取母液0.5~2.0ml于50ml容量瓶中,加碘化钾浓度为0.5~1%,加入10%盐酸溶液,沸水浴中加热约10min(温度升到约80~90℃),冷却后,加抗坏血酸到浓度为0.2~0.5%后以10%的盐酸溶液定容,测定。5.3.2.2曲线的制作:分取标准溶液(1.0μg/ml)0.00/0.25/0.50/0.75/1.00/1.25ml于一组50ml容量瓶中,以下按4.3.2.1步骤操作。5.3.3锡的测定:5.3.3.1分取母液0.5~2.0ml于50ml容量瓶中,以PH3.7的醋酸-醋酸钠溶液定容,测定。5.3.3.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.3.1步骤操作。5.3.4锑的测定:5.3.4.1分取母液0.5~2.0ml于50ml容量瓶中,加碘化钾浓度为0.5~1%,加入10%盐酸溶液,放置1~2min,加抗坏血酸到浓度为0.2~0.5%后以10%的盐酸溶液定容,测定。5.3.4.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.4.1步骤操作。 5.3.5铋的测定:5.3.5.1分取母液0.5~2.0ml于50ml容量瓶中,加入醋酸溶液1.0ml,以20%(V/V) 的盐酸定容。5.3.5.2曲线的制作:分取标准溶液(2.0μg/ml)0.00/0.50/1.00/1.50/2.00/2.50ml于一组50ml容量瓶中,以下按4.3.5.1步骤操作。5.3.6铅的测定:5.3.6.1分取母液0.5~2.0ml于50ml容量瓶中,加入过氧化氢溶液0.5~1.0ml,醋酸溶液1.0~2.0ml,以水定容。5.3.6.2分取标准溶液(1.0μg/ml)0.00/0.25/0.50/0.75/1.00/1.25ml于一组50ml容量瓶中,以下按4.3.6.1步骤操作。六、 分析结果的计算:按下式计算砷锡锑铅铋的百分含量:As、Sn、Sb、Pb、Bi(%)=m1/m0r ×100试中 m1------------从工作曲线上查出的砷锡锑铅铋量(g)m0------------试样量(g)r---------试液分取比
[list][*] [list][*]①实验原理:试样消解后,在盐酸介质中,将试样中的六价硒还原成四价硒,用硼氢化钠或硼氢化钾作还原剂,将四价硒在盐酸介质中还原成硒化氢,由载气(氩气)带入原子化器中进行原子化,在硒空心阴极灯照射下,基态硒原子被激发至高能态,在去活化回到基态时,发射出特征波长的荧光,其荧光强度与硒含量成正比,外标法定量。 [/list][list][*]②试剂仪器:盐酸、硝酸、高氯酸、硼氢化钠、氢氧化钠、硒标准溶液等;原子荧光光谱仪(配硒空心阴极灯)、电热板、微波消解系统等。 硼氢化钠碱溶液:即硼氢化钠溶于氢氧化钠。[/list][list][*]③分析步骤:试样制备,试样消解,设置仪器条件:负高压、灯电流、原子化温度、载气流速等,制备并测定标准系列溶液从而绘制标准曲线,测定试样溶液及试剂空白溶液。 测定之前,需以盐酸溶液为载流,硼氢化钠碱溶液为还原剂,连续用标准系列的零管进样,待读数稳定。 [/list][list][*]④结果计算:试样溶液硒的浓度减去试剂空白溶液硒的浓度,乘以定容体积即为硒的质量,除以样品质量即为硒的含量,若试样溶液稀释则乘以相应的稀释倍数。 [/list] [/list]
我现在用《药典》上的方法做砷,可是发现空白很高,而且测标线和样品时吸光值很不问,跳动幅度很大,即使测同一个样品时,相对偏差有时也会跳到17%,大家能不能帮我分析一下原因,谢谢。砷的测定(氢化物法)测定条件:采用适宜的氢化物装置,以含1%硼氢化钠和0.3%氢氧化钠溶液作为还原剂,盐酸溶液(1—100)为载液,氮气为载气,检测波长为193.7nm,背景校正为氘灯或赛满效应。标准曲线制备:分别精密量取砷标准溶液适量,用2%硝酸溶液稀释制成每1ml分别含砷0、5、10、20、30、40ng的溶液。分别精密量取10ml,置25ml容量瓶中,加25%碘化钾溶液1ml,摇匀,加10%抗坏血酸溶液1ml,摇匀,用盐酸溶液(20—100)稀释至刻度,摇匀,密塞,置80℃水浴中加热3min,取出,放冷。供试品溶液的制备:同铅测定项目的制备一致(用湿消解)测定法 精密吸取空白溶液与供试品溶液各10ml,照标准曲线的制备项下,自“加25%碘化钾溶液1ml”起,依法测定。从标准曲线上读出供试品溶液中砷(As)的含量,计算即得。
最近在用湿法消化处理氢化油,但效果不好,大家一般都采用什么混酸体系呢?如果用硫酸和硝酸处理的话,效果还行,但用硫酸的话,对原吸仪器会不会有什么影响?会不会燃烧后堵了燃烧缝,因为有些硫酸盐是难溶物。我是测镍的
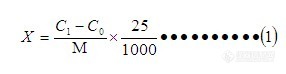
一、摘要目的:寻找一种比较简捷的测定大米中总砷的方法;方法:样品灰化后酸溶解加还原剂,氢化物原子荧光法测定。线性范围在0——100ng/mL范围内遵守郎伯—比尔定律,R值可达到0.9995以上,取样1g左右,定容至25mL检出限为0.01ng/mL,RSD<4.50%,回收率93%~~102%,此方法简便、快速、准确、稳定。二、原理试样经干灰化后,用一定浓度的算溶解置于容量瓶后,加入硫脲使As5+ 预还原为As3+ ,再加入硼氢化钠或硼氢化钾使还原生成砷化氢,由氩气载入石英原子化器中分解为原子态砷,在特制砷空心阴极灯的发射光激发下产生原子荧光,其荧光强度在固定条件下与被测溶液中的砷浓度成正比,与标准系列比较定量。三、试剂1.还原剂:1.5%NaBH4 溶于0.5%NaOH中;2.载液:2%HCL;3.硫脲溶液:(50g/L);4.硫酸溶液(1+9):量取浓硫酸100 mL,小心倒入900 mL超纯水中,混匀;5.标样酸度:5-10(V/V)HCL;6.砷标准溶液:i. 砷标准储备液:(1000μg/mL)国家标准物质中心;http://ng1.17img.cn/bbsfiles/images/2013/07/201307162203_451745_2015870_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/07/201307162205_451746_2015870_3.jpgii. 砷使用标准液:含砷10ng/mL。吸取1.00 mL砷标准储备液于10[
氢化物发生器一般能用那些酸,那些酸不能用,我听别人说氢氟酸不能用,说会减少氢化物器的寿命,别的酸可以用,硝酸,高氯酸,盐酸都可以用,大家认为呢
求助,哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做确认,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰面积峰形都相近,RSD很小,但是随行的对照品溶液在进了6针样品后峰形变矮且拖尾,峰面积也差了好几倍,有做过的这样的吗?
氢化物发生原子吸收法测定电镀废水中砷含量的研究1. 实验部分 1.1 实验仪器及仪器条件AA-800原子吸收光谱仪(美国 PerkinElmer公司),MCA-101微型化学原子化器,T 形石英管:管长140 mm ,内径7 mm ,支管长80 mm ,内径3 mm。砷无极放电灯(美国 PerkinElmer公司),Synery UV 超纯水系统(美国Millipore公司)。分析线波长193.7 nm,灯电流380 mA,光谱通带为0.2 nm,载气流速0.8 L/min,泵速为65 r/min,读数时间15 s,测量方式:标准曲线法,读数方式:峰面积。1.2 主要试剂砷标准溶液:1000 μg/mL,国家钢铁材料测试中心钢铁研究总院,使用时逐级稀释成0. 5 mg/L。盐酸(优级纯,广州化学试剂厂),硼氢化钠(分析纯,天津市福晨化学试剂厂),氢氧化钠(分析纯,广州化学试剂厂),抗坏血酸(分析纯,广州化学试剂厂), 硫脲(分析纯,广州化学试剂厂)。15 g/L硼氢化钠溶液(含10 g/L的NaOH 溶液): 称取NaBH4 1.5 g和NaOH 1.0 g于烧杯中,加100 mL水溶解,现用现配。所有用到的器皿在使用前用15%硝酸浸泡24小时;实验所用水均为超纯水。2.方法原理试样在预还原剂抗坏血酸-硫脲的作用下使五价砷还原为三价砷,进一步由硼氢化钠在酸性溶液中产生的新生态氢,将三价砷转变为砷代氢气体,由氩气载入T型管中,受热分解为原子态砷蒸气吸收波长193.7nm的共振线,其吸收量与砷含量符合朗伯—比尔定律。 3. 试验步骤3.1 标准工作曲线从1000 μg/mL的砷标准溶液中吸取1mL到100 mL容量瓶中,加体积分数为50%的盐酸溶液20 mL,用超纯水定容至刻度,得到一级储备液。浓度为10μg/mL。然后再从一级储备液中吸取1mL至另一100mL容量瓶中,加入体积分数为50%的盐酸溶液20 mL,加入硫脲(50 g/L) +抗坏血酸(50 g/L)混合溶液10 mL,用超纯水定容至刻度。摇匀放入沸水浴中[font=Times New
氢化物消除干扰的途径 在干扰的类型及机理确定以后,即可采取相应的措施来消除和降低干扰,消除分析中常见的干扰,可以通过以下途径。 (一)选择最佳酸度介质 因为氢化物的反应是在酸性介质中进行,因此有关研究报千都要涉及反应液介质问题,通常采用盐酸介质,其浓度对砷、锑、铋的影响不大,允许浓度范围较宽。但随酸的性质不同而有不同的影响,如硝酸对砷和硒的测定有干扰,酸的浓度对硒和碲的测定有影响,要有较高的酸度,而锗、锡、铅的氢化物发生都要求在较严格折浓度下进行测定。 表一 氢化物反应酸度条件 元素价态反应介质元素价态反应介质As +3 1-6NHCl Sb +3 1-6NHCl Te +4 4-6NHCl Bi +3 1-6NHCl Ge +4 20%H3PO4 Se +2 1-6NHCl Sn +4 酒石酸缓冲溶液PH=1.3 控制酸度也可以控制价态干扰,例如在PH=4个时,只有三价砷能转化为砷化氢,而当PH=5时,三价砷和五价砷均可还原成砷化氢,则可测出两种价态砷的总量。 (二)选择最佳还原 剂及用量 有人对比采用Zn和硼氢化钠两种还原体系时,不同离子的干扰效应,发现某些离子的干扰程度取决于所采用的还原剂种类。他们还研究了测定砷和锑时碘化钾对共存离子的掩敝作用,掩敝效果与硼氢化钠的浓度有极密切的关系,总的来说,低的硼氢化钠溶液可以降低共存离子的干扰,原因在于稀溶液不能将金属离子还原成金属。需要指出的是,硼氢化钠溶液不能消除其它可形成氢化物元素的干扰。
求助,有哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做这个,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰形和峰面积都很相近,但是在进样6针样品后的随行对照品溶液,12-羟基硬脂酸甲酯峰的峰形变矮且拖尾,峰面积也变小了一半多,更换了衬管后刚开始峰面积正常,进了样品后就不行了,有什么解决方法吗
根据中国药典(2010版)药品中的砷(As)和汞(Hg)需要采用氢化物原子吸收法进行测试,氢化物发生装置最近才有机会尝试了一下,没啥经验,就将实验过程与结果与大家分享一下,希望诸位多多指教。1.仪器设备日立ZA3000原子吸收光谱仪(带氢化物发生器)砷元素空心阴极灯:北京有色金属研究院国产灯汞元素空心阴极灯:衡水市宁强光源有限公司国产灯Barnstead超纯水装置(美国)2.试剂砷、汞标准溶液:1000μg/mL(国家标准物质中心)硝酸(GR,北京化工厂)、硫酸(AR,北京化工厂)、盐酸(AR,北京化工厂)、高氯酸(AR,北京化工厂)、碘化钾(AR,西陇化工股份有限公司)、高锰酸钾(AR,西陇化工股份有限公司)、盐酸羟胺(AR,西陇化工股份有限公司)、硼氢化钠(国药集团化学试剂有限公司)、氢氧化钠(GR,北京化工厂)实验用水均为去离子水3.砷测试实验(氢化物法)3.1标准溶液的配制砷标准储备液的制备:精密量取砷单元素标准溶液适量,用2%硝酸溶液稀释,制成每1ml含砷(As)0.1μg的溶液。标准曲线的制备:分别精密量取砷标准储备液适量,用2%硝酸溶液稀释制成每1ml分别含砷0ng、5ng、20ng、30ng、50ng的溶液。分别精密量取10ml,置25ml量瓶中,加25%碘化钾溶液(临用前配制)1ml,加10%抗坏血酸溶液(临用前配置)1mL,摇匀,用盐酸溶液(20→100)稀释至刻度,摇匀,密塞,置80℃水浴中加热3分钟,取出,放冷。吸入氢化物发生装置,测定吸收值,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。3.2仪器工作条件仪器条件测定参数元素As原子化器:标准燃烧头测定模式:工作曲线仪器:ZA3000火焰:空气-乙炔信号模式:BKG校正原子化方式:燃气流速:1.2L/min曲线类型:线性检测波长:193.7nm助燃气:160KPa 15L/min计算方式:积分狭缝宽度:1.3nm燃烧头高度:7.5mm延迟时间:0s 灯电流:8mA时间常数:1.0s计算时间:3.0s3.3标准曲线的绘制采用氢化物发生装置,以含1%硼氢化钠和0.4%氢氧化钠溶液(临用前配制)作为还原剂,盐酸溶液(1→100)为载液,氩气为载体,检测波长为193.7nm绘制标准曲线。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311656_530662_2846602_3.bmp3.4供试品溶液的制备与测试取供试品1.0g,精密称定,置锥形瓶中,加硝酸-高氯酸(5:1)混合溶液6ml,混匀,瓶口加一小漏斗,浸泡过夜。置电炉上加热消解,保持微沸,变棕黑色,分批次共加硝酸16ml,持续加热直至溶液澄明,直至白烟散尽,消解液呈淡黄色,放冷,转入25ml量瓶中,用2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀。分别精密量取10ml,置25ml量瓶中,加25%碘化钾溶液(临用前配制)1ml,加10%抗坏血酸溶液(临用前配置)1mL,摇匀,用盐酸溶液(20→100)稀释至刻度,摇匀,密塞,置80℃水浴中加热3分钟,取出,放冷。同法同时制备试剂空白溶液。供试品测试:照标准曲线的制备方法测定吸光度,从标准曲线上读出供试品溶液中砷的含量。3.5测试结果砷元素测试结http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif称样量(g)定容体积(ml)稀释倍数测试值ug/lAs含量mg/kg1031.0060252.50.9940.0621041.0107252.51.0330.0641051.0066252.5-0.275未检出4.汞测试实验(冷吸收法)4.1标准溶液的配制汞标准储备液的制备:精密量取汞单元素标准溶液适量,用2%硝酸溶液稀释,制成每1ml含汞(Hg)1μg的溶液,即得(0~5℃贮存)。4.2.5.3标准曲线的制备:分别精密量取汞标准储备液0ml、0.1ml、0.3ml、0.5ml、0.7ml、0.9ml、1.0ml,置50ml量瓶中,加20%硫酸溶液10ml、5%高锰酸钾溶液0.5ml,摇匀,滴加5%盐酸羟胺溶液至紫红色恰消失,用水稀释至刻度,摇匀。取适量,吸入氢化物发生装置,测定吸收值,以峰面积(或吸光度)为纵坐标,浓度为横坐标,绘制标准曲线。4.2仪器工作条件仪器条件测定参数元素Hg原子化器:汞池测定模式:工作曲线仪器:ZA3000火焰:空气信号模式:BKG校正原子化方式:冷吸收燃气流速:1.2L/min[
近日测陶瓷制品的 砷样品用 4%乙酸浸泡,室温,24h取浸泡液 20mL,加入 (1+1)HCl 5mL,5%硫脲-5%Vc 10mL用水定容至 50mL仪器:海光 AFS-230E(手动进样)载流 5%HCl,还原剂 2%KBH4 + 0.5%NaOH载气400, 屏蔽气1000,断续流动程序使用仪器默认值但是在样品进入反应环之后,产生大量泡沫,废液来不及排走,泡沫竟然充满一级气液分离器之后,连二级气液分离器(水封)也充满了,为了避免水汽冲上原子化器,只好不停用针筒从水封中抽掉废液……汗啊-_-!!这是我第一次做含有乙酸的样品,泡沫超多,而且产生得很快,来不及排走。而以前做过的样品中只有盐酸,却不会产生泡沫,废液都能及时排走。请问哪位前辈 可以解释一下,乙酸对生成氢化物反应的过程会产生哪些影响??怎样才可以减少泡沫产生?
最近我们实验室在测试砷的时候出现砷不稳定的情况?请问各位遇到这种问题是如何解决的呢?大家帮忙看看,是不是测试方法出了什么问题:我的测试方法是:载液:1%的盐酸 1.5%的硼氢化钾标液的配置方法如下:分别量取0,0.5,1,1.5,2.0ML的1000PPB的砷的标准溶液于五个烧杯中,加入30ML左右的水,分别加入10ML盐酸,再分别加入0.4G碘化钾,溶化后分别加入0.2G抗坏血酸,在80度的水域中水浴15分钟,冷却后转移到100ML容量瓶中,定容。上机测试的过程中发现曲线做的很好,但是随着时间的推移,用20PPB的标样做QC,发现20PPB的测试结果不断上升,可以达到30PPB备注:我们实验室只有AAS,没有AFS,所以只有采取用氢化物侧砷的方法仪器预热时间够长,神灯预热时间也很长砷灯是刚刚换的新的大家帮忙看看是不是测试方法出了什么问题?
1.原理食品中的砷可能以不同的化学形式存在,包括无机砷和有机砷。在6mol/L盐酸水浴条件下,无机砷以氯化物形式被提取,实现无机砷和有机砷的分离。在2mol/L盐酸下测定总无机砷。2.试剂盐酸溶液(1+1,量取250mL盐酸,慢慢倒入250mL水中,混匀)、2g/L氢氧化钾溶液(称取氢氧化钾2g溶于水中,稀释至1000mL)、7g/L鹏氢化钾溶液(称取硼氢化钾3.5g溶于500mL 2g/L氢氧化钾溶液中)、100g/L碘化钾-50g/L硫脲混合溶液(称取碘化钾10g、硫脲5g溶于水中,并稀释至100mL混匀)。三价砷(As3+)标准溶液:准确称取三氧化二砷0.1320g,加100g/L氢氧化钾lmL和少量亚沸蒸馏水溶解,转入100mL容量瓶中定容。此标准溶液含三价砷(As3+)lmg/mL。使用时用水逐级稀释至标准使用液浓度为三价砷(As3+)1μg/mL。冰箱保存可使用7d。3.仪器玻璃仪器(使用前经15 %硝酸浸泡24h)、原子荧光光度计、恒温水浴锅。4.分析步骤(1)试样处理①固体试样 称取经粉碎过80目筛的于样2.50g(称样量依据试样含量酌情增减)于25mL具塞刻度试管中,加盐酸(1+1)溶液20mL,混匀,或称取鲜样5.00g(试样应先打成匀浆)于25mL具塞刻度试管中,加5mL盐酸,并用盐酸(1+1)溶液稀释至刻度,混匀。置于60℃水浴锅18h,期间多次振摇,使试样充分浸提。取出冷却,脱脂棉过滤,取4mL滤液于10mI。容量瓶中,加碘化钾一硫脲混合溶液lmL,正辛醇(消泡剂)8滴,加水定容。放置10min后测试样中无机砷。如混浊,再次过滤后测定。同时做试剂空白试验。注:试样浸提冷却后,过滤前用盐酸(1+1)溶液定容至25mL。②液体试样取4mL试样于10mI.容量瓶中,加盐酸(1+1)溶液4mL,碘化钾一硫脲混合溶液lmL,正辛醇8滴,定容混匀,测定试样中总无机砷。同时做试剂空白试验。(2)仪器参考操作条件光电倍增管(PMT)负高压340V;砷空心阴极灯电流40mA;原子化器高度9mm;载气流速600mL/min读数延迟时间2s;读数时间12s;读数方式峰面积;标液或试样加入体积0.5mL。(3)标准系列 无机砷测定标准系列:分别准确吸取lμg/mL三价砷(As3+)标准使用液0、0.05mL、0.1mL、0.25mL、0.5mL、1.0mL于10mL容量瓶中,分别加盐酸(1+1)溶液4mL,碘化钾一硫脲混合溶液1mL,正辛醇8滴,定容。5.结果计算试样中无机砷含量按下式进行计算。X= /式中,X为试样中无机砷含量,mg/kg或mg/L;C1为试样测定液中无机砷浓度,ng/mL;C2为试剂空白浓度,ng/mL;m为试样质量或体积,g或mL;F为固体试样(F= 10mL×25mL/4mL)或液体试样(F=10mL)。
采用铁氰化钾-柠檬酸体系,以氢化物-原子荧光法检测水中铅,研究了最佳测量条件,结果表明,铅的测定范围0.2-20ug/L,标准曲线回归方程IF=134.7×C+9.3,相关系数r=0.9999,检出限0.043ug/L, 线性范围0.2-50ug/L,精密度和准确度均令人满意。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=185392]鉛的測定02.pdf[/url]想改为第2届网络大赛原创作品怎么改呀,搂主可以改一下吗?
EDL=无极放电灯; HCL=空心阴极灯试 剂盐酸,32%(W/V)和37% (W/V)硝酸,65%(W/V)碘化钾 ﹑氢氧化钠﹑硼氢化钠。酸类,碘化钾和氢氧化钠是分析纯;硼氢化钠是试剂纯。在所有试剂中使用了3%(W/V) 硼氢化钠溶液作为还原剂。新配制的溶液使用前过滤。为了一致性在一个系列中对所有试样、标准和空白溶液必须作相同的予处理。尽管由于标准溶液不需要予还原,如果已经选定适当的价态,即使不存在着由碘化钾或高酸度所产生的显著影响,也需采用相同的还原程序。在痕量和超痕量分析中试样与标准溶液具有相同的或很相似的化学组成一般是有利的。对于湖水及饮用水式样下列予还原方法被认为最佳以保证全部还原至较低价态。锑的处理将4ml盐酸[32%(W/V)]和2ml碘化钾[10%(W/V)]溶液加入40ml水样,标准溶液和空白溶液中。溶液即可进行测定,因为还原至锑(III)是瞬时的。为了防止在酸性溶液中碘的生成加入碘化钾后应立即进行测定,而且当碘的浓度较高时将对氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]产生干扰。砷的处理将3ml盐酸[32%(W/V)]和1ml碘化钾[10%(W/V)]溶液加入10ml水样,标准溶液和空白溶液中。在室温下砷(V)还原至砷(III)约需一小时,然后将溶液用于测定。铋的处理在天然水样中由于铋只以III价态存在,因此不需要予还原。于10ml水样,标准溶液和空白溶液中加入0.5ml盐酸[32%(W/V)]此溶液即可进行分析。硒的处理硒(IV)的选择测定将2ml盐酸[32%(W/V)] 加入50ml水样,标准溶液和空白溶液中,可立即进行分析。用此予处理法硒(IV)不被测定。总硒量Se(IV)和Se(VI)]的测定:取25ml水样、准溶液和空白溶液连同20ml盐酸(37%)置于高压釜(Auto-Clave3)中。在电热板上将密闭体系中的混合物加热至80°C,同时搅拌并延续10分钟,当溶液冷却至室温后溶液即可进行分析。碲的处理碲(IV)的选择测定,将1ml盐酸[32%(W/V)] 加入0.5ml水样,标准溶液和空白溶液中,可立即进行分析。用此予处理法碲(IV)不被测定。总碲量[Te(IV)和Te(VI)]的测定:在锥形瓶中将20ml盐酸[37%(W/V)] 加入到20ml水样、标准溶液和空白溶液中并加热至沸2分钟,冷却至室温后溶液即可进行分析。结果及讨论对下列水样进行5种生成氢化物元素的分析:1.Constance湖的表面水2.Constance湖60米深处的水3.Constance湖的纯化饮用水锑的测定用氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定锑,价态对灵敏度的影响[10]。锑(III)的灵敏度以峰值表示约为锑(V)的两倍,因此如果在试样和标准溶液中含有不同价态的锑将产生显著的误差。然而,在酸溶液中加入碘化钾即可使锑(V)还原至锑(III),用于水的分析两个优点,即可确保试样溶液中只有同样的价态,又可使灵敏度对锑(III)增加一倍。但是,如前所述很重要一点是当碘化钾加入后,溶液应立即进行测定,以防止酸溶液中由于过量碘的生成将干扰氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]。从SbCl3和SbCl5配置标准溶液以证明不同价态的锑具有不同灵敏度。用1ml[32%(W/V)]的盐酸直接酸化后以及用KI经过予还原的溶液测定后。两种氧化态之间灵敏度的差别以及用KI予还原后这种差别的消失都清楚的展现在记录仪的波形上。如提及保持不变,KI对已经是三价的锑没有影响。从Constance湖取的水样中用和不用KI子还原测定锑,从记录仪描绘的波形看,显然在这些水样中锑是以5价形式存在,表面水中测定的浓度为0.14ug/l, 60米深处的水为0.12ug/l,纯化饮用水为0.12ug/l。砷的测定对于砷两种氧化态灵敏度的差别不如锑那么明显,砷(V)的灵敏度以峰值表示比砷(III)大约低25%。如果试样中的价态不知道,而标准溶液中的砷是以不同价态的形式存在,最可能导致错误的结果。以下列四种不同砷化合物配置溶液来证明砷(III)和砷(V)之间灵敏度的差别:1.1 g/l AS (V) 用As2O5配制2.1 g/l AS (V) 用Na2HAsO47H2O配制3.1 g/l AS(III) 用Na2AsO2配制4.1 g/l AS(III) 用As2 O3配制所有溶液进一步按1:1000稀释配成As 1mg/l的标准溶液,取50ul相当于50ng砷加入到装着10ml去离子水和1ml盐酸[32%(W/V)]的氢化物反应瓶并用氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定,经KI予还原后按上述进行测定。砷(III)和砷(V)化合物经过KI予还原后得到相同的灵敏度,而与原始价态无关。经还原后与还原前的砷(III) 化合物的灵敏度不等。其原因在于测定所用溶液体积不相同;没有还原的体积为11ml(10ml试样+1ml盐酸)和经予还原的体积为16.5ml(10ml试样+3ml盐酸+1mlKI溶液+2.5ml去离子水冲洗反应瓶)。已知在氢化法中随着体积的增加绝对灵敏度有下降。因此对于试样、空白溶液和标准溶液不仅所用的酸和其它试剂的浓度要相同,而且最后的体积也要一致,这是很重要的。因为砷(III)和砷(V)只见灵敏度有明显差别,对水样、空白溶液和标准溶液都要进行予还原以避免误差。对于从只含砷(III)的贮备液配置的标准溶液可以省略予还原。因为对KI溶液或较高酸度都未发现有明显得影响。从Constance湖的三种水样包括饮用水按上述予还原后测定砷的结果:表面水为1.25ugAs/l,60米深处的水为2.0 μgAs/l,而饮用水为1.9 µ gAs/l。铋的测定可以假定在天然水中铋只以三价形式存在,只有几种已知的不稳定铋酸盐和五氧化铋是以五价形式存在,据我们所知化学实际厂商不能提供稳定的Bi(V)化合物。对于铋的测定时试样只要求进行酸化。所有研究的水样中铋含量全低于0.2ug/l。硒的测定硒(IV)和硒(VI)之间的灵敏度差别较之锑或砷的两种价态间的差别更显著,因此为了测定的准确度应了解试样中硒的价态并把硒(VI)还原为硒(IV)是很重要的。分别用SeO2和H2SeO4配制标准储备液,配成含1g Se(IV)/l的10%(W/V)盐酸溶液和含1.34g Se(VI)/l的水溶液。只有硒(IV)能快速地和可重复地还原为氢化物气体。相反,硒(VI)还原如此的缓慢(即有也很少),因此不能在这种条件下测其峰值或峰面积,要进行准确的测定还原式样中的硒(VI)是很重要的。为此使用各种还原剂如甲醛、甲酸、肼或羟氨进行若干此试验,没有哪一个得到满意的结果。Brodie[1],Catter[8]和Reichert[1]提出用热的4-6M盐酸予还原。Catter找出盐酸浓度和煮沸时间与还原效率之间的关系。在高酸度和敞口瓶中煮沸试样将增加Se(IV)和Se(VII)地逸失。Reichert提出将25ml水样与25ml盐酸(37%)一起煮沸的方法致使1000ngSe损失达15%。由于Constance湖水硒含量典型地低于1ug/l,用相应较低硒量进行重复试验。在敞口烧瓶中将5ng Se(IV)相当于0.2ug Se/l和6.5ng Se(VI)相当于0.26ug/l Se加热至沸;硒的损失高达50%。所有试样在密闭体系中加热以防损失。使用高压釜3可以保证全部还原,回收率为95-100%。在以后的工作中发现只要使用普通玻璃器械用来煮沸溶液硒就有损失。似乎是由于玻璃的表面效应所致。如果用聚乙烯或聚四氟乙烯(PTFE)敞口烧瓶,煮沸时没有发现损失。100ngSe峰值是有差别的,部分是由于部分的体积不同(16和45ml)以及所用酸浓度的不同(分别为0.3M和0.5M)。正如前面所述,对砷的测定,对硒的测定最后体积较大责导致灵敏度稍有下降。还有算浓度太高,达16%(W/V),也引起灵敏度下降。由于硒的两种价态之间灵敏度相差悬殊和Se(VI)不易被还原至Se(IV),因此可以选择区分两种价态。如试样仅用盐酸酸化后分析,则可选择的测定Se(VI)的含量。从硒总量中减去这个数值即可得到试样中Se(VI)的含量。碲的测定碲的两种价态在灵敏度方面的差异与硒很相似,这意味着不能从硼氢化钠作还原剂的氢化物法有效的测定碲(VI),可能此两种元素的高氧化态还原太慢以致难于测定。与硒(VI)相似,用高浓度的盐酸煮沸可以使Te(VI)还原至Te(IV)。与硒相反,在敞口锥形瓶中加热没有损失。沸腾2分钟足以使所有Te(VI)还原至Te(IV)。从两种氧化的碲在煮沸盐酸中予还原和不经予还原的记录波形上看。分别用TeCl4和Te(OH)6整治含量均为1gTe/l的储备液。Te(IV)为3%HCl(W/V)溶液Te(VI)为水溶液。用适当的酸进行稀释配成标准溶液。记录仪波形表明体积大小的差别和酸浓度的差别都不影响灵敏度有任何明显的差别。与硒一样,用氢化法可以选择区分碲的两种价态。在酸化的试样溶液中单独地测定碲(IV)而用盐酸经煮沸后测碲总量。从两数值之差计算出碲(VI)。在水样中Te(IV)总碲二者测定。在任何水样中都没有可检信号出现,意味着Te(IV)含量低于0.01ug/l,碲总量低于0。025ug/l。实际上根据检出限的差别对Te(IV)可以用50ml水样为最大允许量,但是对总碲只能用20ml与20ml盐酸混合。联系人:展高举 13611340354 zhangaoju@yahoo.com.cn
【原创】植物奶油在中国普遍使用 或酿食物史上最大灾难http://bbs.instrument.com.cn/shtml/20101108/2911265/【原创】植物奶油,美味还是祸端?http://bbs.instrument.com.cn/shtml/20101123/2947740/【转帖】你今天吃反式脂肪了么——令你惊讶的食物营养真相http://bbs.instrument.com.cn/shtml/20101122/2944528/【讨论】植物奶油之专题贴http://bbs.instrument.com.cn/shtml/20101110/2916615/【讨论】卫生部正评估植物奶油风险http://bbs.instrument.com.cn/shtml/20101110/2916010/主题:【讨论】奶酪、芝士、牛油、黄油,你吃吗?http://bbs.instrument.com.cn/shtml/20101113/2923168/主题:【原创】【第三届原创征集】测测你身边的氢化油(new!)http://bbs.instrument.com.cn/shtml/20101115/2926677/主题:【原创】带你了解“植物奶油”(氢化油)http://bbs.instrument.com.cn/shtml/20101108/2911426/主题:【讨论】氢化植物油不等于反式脂肪酸http://bbs.instrument.com.cn/shtml/20101115/2926383/主题:【转帖】反式脂肪酸有安全风险 国内缺乏食用标准http://bbs.instrument.com.cn/shtml/20101130/2965242/主题:【分享】反式脂肪酸是何方“妖孽”http://bbs.instrument.com.cn/shtml/20100822/2735128/主题:【讨论】营养标签中的“反式脂肪酸”http://bbs.instrument.com.cn/shtml/20100617/2618310/主题:【简讯】反式脂肪酸检测方法通过国家食品工业标准委员会专家审定http://bbs.instrument.com.cn/shtml/20080123/1144896/主题:【转帖】麦当劳所售的油炸薯条反式脂肪酸含量超标http://bbs.instrument.com.cn/shtml/20080303/1178170/
有没有做过原子吸收氢化物发生器测砷的朋友,有几个问题弄不明白,求解答1、消解后赶酸不赶酸?赶酸温度是多少?不赶酸的话对加热用的石英管有没有影响?我没赶酸,石英管易出现白色结晶斑点,清洗不掉的2、氢化物测砷,消解之后定容之前,要不要加VC、硫脲、碘化钾把五价砷还原为三价砷3、氢化物测砷,标准曲线浓度点最大取多少,对应的A是多少?我最大取10ppb,但是吸收值上不去,曲线做不好
。锑的处理将4ml盐酸[32%(W/V)]和2ml碘化钾[10%(W/V)]溶液加入40ml水样,标准溶液和空白溶液中。溶液即可进行测定,因为还原至锑(III)是瞬时的。为了防止在酸性溶液中碘的生成加入碘化钾后应立即进行测定,而且当碘的浓度较高时将对氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]产生干扰。砷的处理将3ml盐酸[32%(W/V)]和1ml碘化钾[10%(W/V)]溶液加入10ml水样,标准溶液和空白溶液中。在室温下砷(V)还原至砷(III)约需一小时,然后将溶液用于测定。铋的处理在天然水样中由于铋只以III价态存在,因此不需要予还原。于10ml水样,标准溶液和空白溶液中加入0.5ml盐酸[32%(W/V)]此溶液即可进行分析。硒的处理硒(IV)的选择测定将2ml盐酸[32%(W/V)] 加入50ml水样,标准溶液和空白溶液中,可立即进行分析。用此予处理法硒(IV)不被测定。总硒量Se(IV)和Se(VI)]的测定:取25ml水样、准溶液和空白溶液连同20ml盐酸(37%)置于高压釜(Auto-Clave3)中。在电热板上将密闭体系中的混合物加热至80°C,同时搅拌并延续10分钟,当溶液冷却至室温后溶液即可进行分析。碲的处理碲(IV)的选择测定,将1ml盐酸[32%(W/V)] 加入0.5ml水样,标准溶液和空白溶液中,可立即进行分析。用此予处理法碲(IV)不被测定。总碲量[Te(IV)和Te(VI)]的测定:在锥形瓶中将20ml盐酸[37%(W/V)] 加入到20ml水样、标准溶液和空白溶液中并加热至沸2分钟,冷却至室温后溶液即可进行分析。结果及讨论对下列水样进行5种生成氢化物元素的分析:1.Constance湖的表面水2.Constance湖60米深处的水3.Constance湖的纯化饮用水锑的测定用氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定锑,价态对灵敏度的影响[10]。锑(III)的灵敏度以峰值表示约为锑(V)的两倍,因此如果在试样和标准溶液中含有不同价态的锑将产生显著的误差。然而,在酸溶液中加入碘化钾即可使锑(V)还原至锑(III),用于水的分析两个优点,即可确保试样溶液中只有同样的价态,又可使灵敏度对锑(III)增加一倍。但是,如前所述很重要一点是当碘化钾加入后,溶液应立即进行测定,以防止酸溶液中由于过量碘的生成将干扰氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]。从SbCl3和SbCl5配置标准溶液以证明不同价态的锑具有不同灵敏度。用1ml[32%(W/V)]的盐酸直接酸化后以及用KI经过予还原的溶液测定后。两种氧化态之间灵敏度的差别以及用KI予还原后这种差别的消失都清楚的展现在记录仪的波形上。如提及保持不变,KI对已经是三价的锑没有影响。从Constance湖取的水样中用和不用KI子还原测定锑,从记录仪描绘的波形看,显然在这些水样中锑是以5价形式存在,表面水中测定的浓度为0.14ug/l, 60米深处的水为0.12ug/l,纯化饮用水为0.12ug/l。砷的测定对于砷两种氧化态灵敏度的差别不如锑那么明显,砷(V)的灵敏度以峰值表示比砷(III)大约低25%。如果试样中的价态不知道,而标准溶液中的砷是以不同价态的形式存在,最可能导致错误的结果。以下列四种不同砷化合物配置溶液来证明砷(III)和砷(V)之间灵敏度的差别:1.1 g/l AS (V) 用As2O5配制2.1 g/l AS (V) 用Na2HAsO47H2O配制3.1 g/l AS(III) 用Na2AsO2配制4.1 g/l AS(III) 用As2 O3配制所有溶液进一步按1:1000稀释配成As 1mg/l的标准溶液,取50ul相当于50ng砷加入到装着10ml去离子水和1ml盐酸[32%(W/V)]的氢化物反应瓶并用氢化物[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法测定,经KI予还原后按上述进行测定。砷(III)和砷(V)化合物经过KI予还原后得到相同的灵敏度,而与原始价态无关。经还原后与还原前的砷(III) 化合物的灵敏度不等。其原因在于测定所用溶液体积不相同;没有还原的体积为11ml(10ml试样+1ml盐酸)和经予还原的体积为16.5ml(10ml试样+3ml盐酸+1mlKI溶液+2.5ml去离子水冲洗反应瓶)。已知在氢化法中随着体积的增加绝对灵敏度有下降。因此对于试样、空白溶液和标准溶液不仅所用的酸和其它试剂的浓度要相同,而且最后的体积也要一致,这是很重要的。因为砷(III)和砷(V)只见灵敏度有明显差别,对水样、空白溶液和标准溶液都要进行予还原以避免误差。对于从只含砷(III)的贮备液配置的标准溶液可以省略予还原。因为对KI溶液或较高酸度都未发现有明显得影响。从Constance湖的三种水样包括饮用水按上述予还原后测定砷的结果:表面水为1.25ugAs/l,60米深处的水为2.0 μgAs/l,而饮用水为1.9 µ gAs/l。铋的测定可以假定在天然水中铋只以三价形式存在,只有几种已知的不稳定铋酸盐和五氧化铋是以五价形式存在,据我们所知化学实际厂商不能提供稳定的Bi(V)化合物。对于铋的测定时试样只要求进行酸化。所有研究的水样中铋含量全低于0.2ug/l。硒的测定硒(IV)和硒(VI)之间的灵敏度差别较之锑或砷的两种价态间的差别更显著,因此为了测定的准确度应了解试样中硒的价态并把硒(VI)还原为硒(IV)是很重要的。分别用SeO2和H2SeO4配制标准储备液,配成含1g Se(IV)/l的10%(W/V)盐酸溶液和含1.34g Se(VI)/l的水溶液。只有硒(IV)能快速地和可重复地还原为氢化物气体。相反,硒(VI)还原如此的缓慢(即有也很少),因此不能在这种条件下测其峰值或峰面积,要进行准确的测定还原式样中的硒(VI)是很重要的。为此使用各种还原剂如甲醛、甲酸、肼或羟氨进行若干此试验,没有哪一个得到满意的结果。Brodie[1],Catter[8]和Reichert[1]提出用热的4-6M盐酸予还原。Catter找出盐酸浓度和煮沸时间与还原效率之间的关系。在高酸度和敞口瓶中煮沸试样将增加Se(IV)和Se(VII)地逸失。Reichert提出将25ml水样与25ml盐酸(37%)一起煮沸的方法致使1000ngSe损失达15%。由于Constance湖水硒含量典型地低于1ug/l,用相应较低硒量进行重复试验。在敞口烧瓶中将5ng Se(IV)相当于0.2ug Se/l和6.5ng Se(VI)相当于0.26ug/l Se加热至沸;硒的损失高达50%。所有试样在密闭体系中加热以防损失。使用高压釜3可以保证全部还原,回收率为95-100%。在以后的工作中发现只要使用普通玻璃器械用来煮沸溶液硒就有损失。似乎是由于玻璃的表面效应所致。如果用聚乙烯或聚四氟乙烯(PTFE)敞口烧瓶,煮沸时没有发现损失。100ngSe峰值是有差别的,部分是由于部分的体积不同(16和45ml)以及所用酸浓度的不同(分别为0.3M和0.5M)。正如前面所述,对砷的测定,对硒的测定最后体积较大责导致灵敏度稍有下降。还有算浓度太高,达16%(W/V),也引起灵敏度下降。由于硒的两种价态之间灵敏度相差悬殊和Se(VI)不易被还原至Se(IV),因此可以选择区分两种价态。如试样仅用盐酸酸化后分析,则可选择的测定Se(VI)的含量。从硒总量中减去这个数值即可得到试样中Se(VI)的含量。碲的测定碲的两种价态在灵敏度方面的差异与硒很相似,这意味着不能从硼氢化钠作还原剂的氢化物法有效的测定碲(VI),可能此两种元素的高氧化态还原太慢以致难于测定。与硒(VI)相似,用高浓度的盐酸煮沸可以使Te(VI)还原至Te(IV)。与硒相反,在敞口锥形瓶中加热没有损失。沸腾2分钟足以使所有Te(VI)还原至Te(IV)。从两种氧化的碲在煮沸盐酸中予还原和不经予还原的记录波形上看。分别用TeCl4和Te(OH)6整治含量均为1gTe/l的储备液。Te(IV)为3%HCl(W/V)溶液Te(VI)为水溶液。用适当的酸进行稀释配成标准溶液。记录仪波形表明体积大小的差别和酸浓度的差别都不影响灵敏度有任何明显的差别。与硒一样,用氢化法可以选择区分碲的两种价态。在酸化的试样溶液中单独地测定碲(IV)而用盐酸经煮沸后测碲总量。从两数值之差计算出碲(VI)。在水样中Te(IV)总碲二者测定。在任何水样中都没有可检信号出现,意味着Te(IV)含量低于0.01ug/l,碲总量低于0。025ug/l。实际上根据检出限的差别对Te(IV)可以用50ml水样为最大允许量,但是对总碲只能用20ml与20ml盐酸混合。结 束 语随着原子序数和金属性质的增加,元素周期表第V A和VI A族元素的较高氧化态其稳定性和还原为氢化物的速度都在减少。意味着高价很容易并快速的还原至低价,到高价还原至氢化物是较慢的。例如,VA族中的砷(原子序数33)在室温和碘化钾存在下,从V价还原至III价约需60分钟,然而同样处理,对锑可立即进行。对铋(原子序数83)已知它的五价稳定化合物尽有几种。而对砷其高价态还原至氢化物比碲快得多。对两种氧化态用峰值差别来表示是很清楚的。对砷约差25%,对锑差50%。
[align=center]曾可明[/align][align=center]湖南省泸溪县疾病预防控制中心(湖南泸溪,416100)[/align] 原子荧光光度计是研究物质生成氢化物后原子化产生的荧光强度测定的仪器。因为要将待测物质变成气态氢化物及新生态原子,所以从仪器结构上讲,就多了氢化物发生装置及原子化器部分。因此,影响氢化物发生原子荧光光度计分析结果的可靠性的因素就会增多。所以用好氢化物发生原子荧光光度计的必须掌握的关键就比较多。本人的使用和实践经验表明,原子荧光大多数出现的不是仪器问题,而是使用问题。本文根据作者多年使用该仪器的实践经验,总结了用好原子荧光光度计的几个最关键的问题。可供同行们参考,希不吝指正。1 空心阴极灯类型的选择 因该方法是测激发荧光强度的大小,故要求高强度灯比普通型的光强度大几十至几百倍,主要用于荧光共振线的测定,以提高测定灵敏度,适于微量元素的分析。2 灯电流的选择 原子荧光灯电流的选择很重要,它直接决定了光源的强度,直接影响仪器的信噪比(或灵敏度)。如果灯电流选择很小,会因光强度小,而使仪器的信噪比会很小,有人认为灯电流小,噪声也小,其实不然。因为原子荧光光度计的噪声包括电噪声和光噪声两部分,其中电噪声是个仪器噪声的主要部分,光噪声一都很小,如果采用脉冲电源,光噪声会更小(大多数原子荧光光度计采用脉冲电源)。以,如果选择的灯电流很小,只能适当降低一些光噪声,但不能降低电噪声,不能真正改仪器的信噪比。但是,灯电流小了以后,光信号将随之变小,使机的信噪比变小。以过小的灯电流是不合适的。如果灯电流选择过大,虽然光信号增大了,但是相应的光噪声增大,还是不能提高仪器的信噪比,甚至会降低仪器的信噪比。因为灯电流大了以后,会使灯发热,造成漂移。因此,选择合适的灯电流,非常重要。灯电流不能太大,不能太小,为了提高灵敏度,较小的灯电流为好。但要于与日盲光电倍增管的负高压和仪器机的噪声综合考。如果光电倍增管选择较低的高压,同时,灯电流要选择得小一点为好。不同厂家生产的不同型号的仪器,其灯电流的选择可能不同。3 负高压的选择 目前国产的原子荧光光度计中,绝大多数都是采用日盲光电倍增管,光电倍增管的负高压大多在200~500V之内。如果仪器的噪声很大,光电倍增管就应选择较低的负高压。不同的厂家生产的不同型号的仪器,其负高压的选择可能不同。总之,应根据待测物质的含量选择调整负高压或灯电流的大小。4 载气流量的选择 载气是将测物还原产生的气态氢化物送入石英原子化器中原子化。如果载气流量过小则不能有效载入,使测定的荧光强度值减小。如果载气流量过大,则产生的氢气稀释气态氢化物,使原子化效率减低,荧光强度值减低,故应选择适宜的载气流量。5 屏蔽气流量的选择 屏蔽气的作用是防止石英原子化器产生的新生态原子被氧化及荧光猝灭。屏蔽气流量过小,不能有效防止氧化及荧光猝灭。如果过高则可稀释氩-氢火焰,使原子化效率降低,荧光强度信号变小。故应适宜选择。6 原子化器高度的选择 原子化器高度的选择应与测定的光源通过原子化器的光斑相重合,以提高原子化效率,过高或过低都会降低原子化效率,使荧光强度信号变低。7 还原剂浓度的选择 在氢化物发生的过程中,还原剂的浓度至关重要。因为它直接决定了产生气态氢化物浓度的大小,它是氢化物发生效率的重要试剂。一要求浓度足量并过量;二要求纯度要高,含干扰杂质少;三要防潮解失效;四要最好现用现配,以防止氧化分解失效。8 载液介质的选择 载液多选择盐酸(或硝酸)为介质,尽量避免使用硫酸和磷酸。其原因是硫酸和磷酸是氧化性酸,不宜用于原子荧光测定的还原体系中。而硝酸除外,因它对测汞的荧光信号增大稍有贡献,常用在测汞体系中。对载液的要求是应选择高纯试剂,这一点显得至关重要。载液如果含有待测元素杂质则测定的标准空白含量会很高,这对测定结果的准确性影响很大。盐酸通常含砷较高,硫酸通常含硒,故要求去砷,去硒处理。应慎重选择高纯可靠的该试剂。总之,要用好原子荧光学问很深,涉及的问题很多。本人认为以上的八个问题是直接影响分析检测数据可靠性最关键的问题,值得深入探讨。不当之处,敬请斧正。 2020.08.21







 我要推广仪器
我要推广仪器
 下载APP
下载APP