江苏出入境检验检疫局动植物与食品检测中心2015年能力验证计划目录 实施机构:江苏出入境检验检疫局动植物与食品检测中心通讯地址:南京市中华路99号 邮编:210001网站:http://www.apfic.com/和http://www.foodmate.net监督电话:025-52345193计划编号计划名称测试项目对应的实验室领域代码对应的PT子领域推荐的测试/测量方法实施时间实施机构及联络信息预计费用APFIC PTP-001 2015果蔬汁中氯氰菊酯、氰戊菊酯和毒死蜱农药残留量的测定氯氰菊酯,氰戊菊酯和毒死蜱227.02食品与农产品中残留物GB/T 5009.146-2003植物性食品中有机氯和拟除虫菊酯类农药多种残留量的测定GB/T 5009.145-2003 植物性食品中有机磷和氨基甲酸酯类农药多种残留的测定2015.8~2015.12联系人:赵增运、沈伟健电话:025-52345193,传真:025-52345180E-mail:shenwj18@jsciq.gov.cn1000元
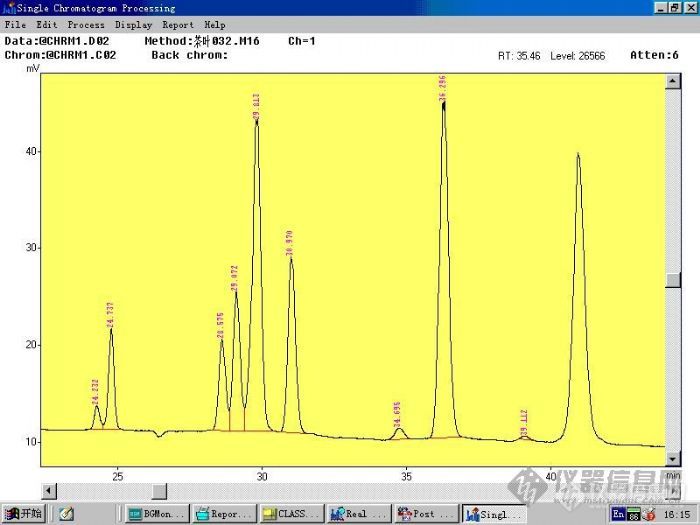
做茶叶农残检验氯氰菊酯和氟氰戊菊酯。氯氰菊酯出3个峰,氟氰戊菊酯出2个峰,氯氰菊酯的第3个峰和氟氰戊菊酯的第1个峰分不开,不知道各位大大有碰到过这样的情况吗?该怎么解决呢?岛津GC-17A,ECD 310℃ inj:280℃ 柱温180℃-2min-10℃/min-280℃-25minflow:2ml/min恒流进样 进样1μl 标样1μg/ml 柱子是兰化的农残1号柱0.32MM*15m[img]http://ng1.17img.cn/bbsfiles/images/2007/12/200712141704_72905_1769105_3.jpg[/img]24.23 min24.73min 氯菊酯 34min 36min 顺式氰戊菊酯 最后1个是溴氰菊酯 28min,29min是氯氰菊酯 29.8的峰没分开 30.9min是氟氰戊菊酯[img]http://ng1.17img.cn/bbsfiles/images/2007/12/200712141704_72906_1769105_3.jpg[/img]粉红的是氯氰菊酯,黑色的是氟氰戊菊酯 这个图是我进高浓度样做的
求助,有做农残[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的老师吗?我们扩GB23200.121氟氰戊菊酯、氰戊菊酯、联苯菊酯、溴氰菊酯这四个菊酯,但是优化不出来。
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测果蔬中菊酯类(氯氰菊酯、氟氯氰菊酯、氯氟氰菊酯,氰戊菊酯,溴氰菊酯)农药,刚开始进标准混合溶液时,它们都有出峰的,可是再进了十几个果蔬样品后,再重新进标准溶液后,质控的标准溶液就不出峰了,也就是说后面出峰的菊酯类走着走着就没有响应了,前面出现出峰的比如有机磷等出峰还是正常的,然后重新老化了VF-1701MS色谱柱,后面出峰的菊酯类标准溶液还是没有出峰?现在要如何解决?
有参加CNAS下半年的茶叶中溴氰菊酯、甲基毒死蜱、甲氰菊酯、硫丹农药残留量的测定能力验证的吗?有做预实验的吗?都是用什么标准检测的?
按NY/T761-2008做蔬菜中菊酯类农药残留(三唑酮、联苯菊酯、甲氰菊酯、三氟氯氰菊酯、氟氯氰菊酯、氯氰菊酯、溴氰菊酯、氰戊菊酯),回收率普遍达到180%,请教各位高手有什么原因造成回收率这么高?
最近我们做蔬菜中菊酯类农药残留(六六六、滴滴涕、氯菊酯、氟氯氰菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯),样品均为未检出,标准曲线的系列浓度为:各农药浓度均为100ng/mL、200ng/mL、400ng/mL、800ng/mL、1000ng/mL,各化合物的线性关系是不错,可是就是样品的农药残留均为未检出,我们也有做加标回收率,问题是加标回收的样品:氯菊酯、氟氯氰菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯这几个菊酯类物质均未出峰,也就是回收率为0啊,我们蔬菜样品的前处理是按照NY/T761-2008来的:称取25.0克蔬菜样品,加入50.0mL乙腈提取,加6克氯化钠使乙腈层与水层分层,取10.0mL乙腈层用旋转蒸发仪于40℃水浴中减压旋转至近干,用正已烷:丙酮=90:10溶解,过艾捷尔的Carb/NH2复合柱(1000mg/6mL)净化除色素,氮吹,用色谱纯正已烷1.0mL定容。可是为什么回收率会为0呢,每进10个样品我们也有插入一个1000ng/mL的标准溶液,标准溶液的测定数据正常啊,为什么就是加标回收的样品不正常?
能力验证名称:茶叶中溴氰菊酯、甲基毒死蜱、甲氰菊酯、硫丹农药残留量的测定编号:CNAS T0616有效期:具体实施时间:2011.6-2011.9组织方:中国测试技术研究院下个星期样品送到
大家做氰戊菊酯标准曲线时,是否发现过会出现氰戊菊酯和顺式氰戊菊酯两个峰,如果我要测这两种农残的含量,应该怎么做?谢谢
蔬菜中氰戊菊酯残留检测中用标样出峰,是出双峰吗?有做农药残留的高手指点下,或加我QQ81314265指导
要用液相测土壤中农残含量。有机磷类:辛硫磷(提取剂:丙酮;定容:正己烷)菊酯类:功夫(高效氯氟氰菊酯)(提取剂:石油醚+丙酮;定容:丙酮)流动相怎么选择?原因?波长怎么选择?根据?其他的条件都怎么确定?新手,不懂,望指教
今天做拟除虫菊酯类农药方法确认,只加了溴氰菊酯和氰戊菊酯结果出了6个峰,一开始以为是杂质,重配后依旧,这正常吗?怎么定量啊?用的hp-5 30*0.32*0.25的柱子,柱温240℃ 进样口270℃ 25Psi 检测器300℃。
有谁参加了这次玉米中的溴氰菊酯、氯氰菊酯残留检测的比对,可以交流一下吗?我们做的初步结果为1号样为100、200ppb,2号样为50、100ppb,但是我们的回收率较高,约为130%,有朋友能交流吗?
一、前言六六六是一类有机氯杀虫剂。是广谱杀虫剂,具有胃毒、触杀和熏蒸三中作用方式。效力强而持久,属高残留农药品种。三唑酮是一种高效、低毒、低残留、持效期长、内吸性强的三唑类杀菌剂。被植物的各部分吸收后,能在植物体内传导。对锈病和白粉病具有预防、铲除、治 疗等作用。对多种作物的病害如玉米圆斑病、麦类云纹病、小麦叶枯 病、凤梨黑腐病、玉米丝黑穗病等均有效。对鱼类及鸟类较安全。对蜜蜂和天敌无害。三唑酮可以与许多杀菌剂、杀虫剂、除草剂等现混现用。三氯杀螨醇也称开乐散,纯品为固体,可做农药,广谱性杀螨剂,对成螨、幼若螨和卵均有效。三氯杀螨醇在酸性中稳定,遇碱易分解。毒性属于低素毒杀螨剂,对人、畜低毒,对多种天敌无害。菊酯属于广谱性杀虫剂,具有速效、高效、低毒、低残留,对作物安全等特点,除对140多种害虫防治有特效外,有些菊酯类农药还对地下害虫和螨类害虫有较好的防治效果。菊酯有天然菊酯及化学合成菊酯。天然菊酯的主要成分为除虫菊素;化学合成的菊酯称为拟除虫菊酯,种类较多,有氯菊酯、胺菊酯、氯氰菊酯、溴氰菊酯、右旋反式烯丙菊酯等,这些成分都属于世界卫生组织推荐可用于防治卫生害虫及其媒介的农药。氨基甲酸酯是重要的药物和农药,广泛应用于农业生产,病虫害防治。
[b][color=#444444]食品中的氯氰菊酯、氰戊菊酯残留量检测需用何种色谱柱?[/color][color=#444444]现GB/T 14929.4—1994和 SN 0217—93的检测方法所要求的色谱柱类型不一样,不知要采用何种色谱柱比较[/color][/b]
最近用的方法检测菊酯类农药氟氰戊菊酯、溴氰菊酯及顺式氰戊菊酯回收率超低的,请问有什么好的方法做比较好
求助啊,我是刚接触农残检测半年不到的小白一枚。请问各位: 我们实验室目前只有GCMS,是新购入的,所以我们的农药统一用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检,其中可能有些是标准里说不适合[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]应该用液相的。然后,有几种农药,我们之前检测能正常出峰,最低出峰浓度可以到0.005ppm水平。但是在大概十天之前,我发现有百菌清、溴氰菊酯、灭多威的出峰都消失了,在0.1ppm也不出峰。想问问各位大神这个问题的原因是什么,然后要怎么解决呐???谢谢各位啦~?
最近发现一个很烦的问题,做茶叶中S-氰戊菊酯定量已经好几年了,最近又做大米农残。发现做过大米样品后,S-氰戊菊酯变成了两个峰,与氰戊菊酯异构体混合物标样出峰几乎一样,都出了两个峰。于是换了新柱子,问题解决了,S-氰戊菊酯峰很好,一个峰。但是昨天刚做了7个大米样,再进标样,发现问题又出了。有什么好办法呢?以前一根柱子仅做茶叶和蔬菜能用好久,现在这一做大米这样了。另外,还发现Lambda-氯氟氰菊酯也存在这个问题。可能大米净化没做彻底也有关系。老化柱子后情况没有一点改善,不知有没有解决办法。菊酯类异构体转换问题大家是否也有遇到过?
各位老师大家好,想请教一个问题,本人参照农业部公告781-9-2006做蜂蜜中的氟胺氰菊酯,标准里是出单峰,但是我做出来好像是双峰,用质谱走也是双峰,我问下各位老师,氟胺氰菊酯到底几个峰?http://ng1.17img.cn/bbsfiles/images/2017/03/201703062357_01_3193564_3.jpg
出口蔬菜中氯菊酯、溴氰菊酯残留量检验方法 中华人民共和国进出口商品检验行业标准 SN 0217-93 Method for the determination of permethrin, cypermethrin, fenvalerate, deltamethrin residues in vegetables for export 1 主题内容与适用范围 本标准规定了出口蔬菜中氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯残留量检验的抽样、制样和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定法。 本标准适用于出口花菜、蘑菇中氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯残留量的检验。2 抽样和制样 2.1 检验批 以不超过 1 000 件为一检验批。 同一检验批的商品应具有相同的特征,如包装、标记、产地、规格、等级等。 2.2 抽样数量 批量,件 最低抽样数,件 1~25 1 26~100 5 101~250 10 251~1 000 152.3 抽样方法 按 2.2条规定的件数随机抽取,逐件开启。每件至少取 500g作为原始样品,原始样 品总量不得少于 4kg,加封后,标明标记并及时送实验室。 2.4 试样制备 分取出部分有代表性样品,取可食部分切碎,用四分法缩分出 1kg左右。置高速组织 捣碎机中,捣碎成果酱状,均分成二份,装入洁净容器内,密封,标明标记。2.5 试样保存 将试样于-18℃以下冷冻保存。 注:在抽样和制样的操作过程中,必须防止样品受到污染和发生残留物含量的变化。 3 测定方法 3.1 方法提要 蔬菜中残留的氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯,采用丙酮提取,提取液用正已烷萃取,硅胶柱净化,二氯甲烷洗脱。净化液用配有电子俘获检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定, 外标法定量。 3.2 试剂和材料 除另有规定外,试剂均为分析纯,水为蒸馏水或相适应的去离子水。 3.2.1 丙酮:重蒸馏。 3.2.2 正已烷:重蒸时收集 68~69℃馏分。 3.2.3 苯:重蒸馏。 3.2.4 二氯甲烷:重蒸馏。 3.2.5 无水硫酸钠: 650℃灼烧 4 h,冷却后贮于密封瓶中备用。 3.2.6 2%(m/m)硫酸钠水溶液:称取 2 g无水硫酸钠,溶于 100 mL水中。 3.2.7 硅胶:Merck Kieselgel60,70~230目。在130℃烘箱中烘 10h,贮于密封瓶中备用。 3.2.8 氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯标准品:纯度 ≥99%。 3.2.9 氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯标准溶液:分别准确称取适量的农药标 准品,用少量苯溶解,然后用正已烷各配制成浓度为 0.100 mg/mL的储备液。根据需要再 稀释配制成适用浓度的混合标准工作液。 3.3 仪器和设备3.3.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]并配备电子俘获检测器。 3.3.2 振荡器。3.3.3 离心管:具塞 50 mL。 3.3.4 微型层析柱:15 cm×0.5 cm(内径),带有 10 mL储液斗。3.3.5 空气流(或氮气)浓缩装置。 3.3.6 刻度试管:10 mL,具磨口塞。3.3.7 无水硫酸钠柱:6 cm×1.8 cm(内径),内装 5 cm高的无水硫酸钠。3.3.8 微量注射器:10μL。3.3.9 脱脂棉:用正已烷回流 2 h,取出挥发至干,保存在清洁容器中备用。3.4 测定步骤 3.4.1 提取 称取约20g试样,精确至0.1g,置于250mL锥形瓶中。加入80mL丙酮,振荡40min,过滤。用丙酮洗涤残渣。将滤液收集在100 mL容量瓶中并以丙酮稀释定容。3.4.2 净化 准确吸取5mL提取液于具塞离心管中,加入20mL硫酸钠水溶液(2%),用正已烷对丙酮-水溶液相萃取二次(每次 10 mL)。合并正已烷萃取液,并通过无水硫酸钠柱脱水,以少量正已烷洗涤柱。将合并的流出液浓缩至约1mL。于微型层析柱的下端填入少量脱脂棉,依次装入0.5cm高的无水硫酸钠、用正已烷拌 湿的1g硅胶和1cm高的无水硫酸钠。用5mL正已烷预淋层析柱,弃去流出液。待液面下降至上层无水硫酸钠表面时,将上述浓缩液倒入柱内,并用5mL正已烷-二氯甲烷(4+1) 洗涤器皿,倒入柱内,弃去流出液。继用二氯甲烷洗脱层析柱,收集洗脱液10mL于收集管 内并加入2mL正已烷。浓缩以除尽二氯甲烷(温度控制于40℃),再以正已烷定容为10mL,供[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。3.4.3 测定 3.4.3.1 色谱条件 a. 色谱柱:石英毛细管柱,5 m×0.53 mm(内径), 2.65 μm (膜厚度)。固定 相为:HP-1[100%dimethyl-polysiloxane (Gum)]。b. 氮气:纯度≥99.99% ,13.3 mL/min。c. 尾吹气:氮气,20 mL/min。d. 柱温:245℃。 e. 进样口温度: 260℃。 f. 检测器温度:280℃。g. 进样方式:柱头进样,不分流。 3.4.3.2 色谱测定 根据试样中被测农药含量情况,选定峰高相近的标准工作液,标准工作液和待测样液中农药的响应值均应在仪器检测的线性范围内。对标准工作液与样液应体积参插进样测定。 在上述色谱条件下,被测农药的保留时间为:氯菊酯约 2.5 min,氯氰菊酯约 3.4.3.3 min, 氰戊菊酯约3.4.3.4 min,溴氰菊酯约 5.7 min。 3.4.4 空白试验:除不称取试样外,均按上述测定步骤进行。3.5 结果的计算和表述 用色谱数据处理机或按下式计算试样中农药的残留含量: Acs X=━━━━━ Asc 式中:X- 试样中被测农药的含量,mg/kg;A-样液中被测农药的色谱峰面积(或峰高),mm**2(mm);As-混合标准工作液中被测农药的色谱峰面积(或峰高),mm**2(mm);Cs-混合标准工作液中被测农药的浓度,μg/mL; c-最终样液所代表的试样浓度,g/mL。 注:计算结果需将空白值扣除。 4 测定低限、回收率 4.1 测定低限 本方法的测定低限:氯菊酯为 0.020 mg/kg,氯氰菊酯为0.040 mg/kg,氰戊菊酯为 0.020 mg/kg,溴氰菊酯为 0.020 mg/kg。4.2 回收率 回收率的实验数据:氯菊酯浓度在0.020~1.00mg/kg范围内,回收率为93.4%~113.8% 氯氰菊酯浓度在 0.040~2.00 mg/kg 范围内,回收率为 91.7%~105.7% ; 氰戊菊酯浓度在0.020~1.00mg/kg范围内,回收率为86.7%~98.6%;溴氰菊酯浓度在 0.020~1.00 mg/kg范围内,回收率为 93.0%~114.2%。 附加说明:本标准由中华人民共和国国家进出口商品检验局提出。 本标准由中华人民共和国上海进出口商品检验局负责起草。本标准主要起草人陈余英、习娟华、朱坚。中华人民共和国国家进出口商品检验局 1993-12-28 批准 1994-05-01实施
各位,我是农药残留的生手,向各位求教:溴氰菊酯在水中和土壤中的残留检测方法以及标准,溴氰菊酯通过呼吸道的急性毒性和对鱼类的毒性试验方面有关的文献,可以在哪里查得到?急用,谢谢了!
谁做过毒死蜱和氯氰菊酯55%乳油混合制剂的农药残留分析?有没有具体的分析方法?
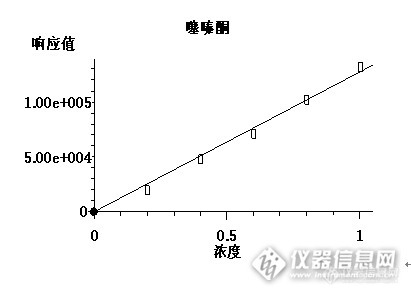
茶叶中硫丹、甲氰菊酯、噻嗪酮类农药残留的检测1、仪器: 安捷伦5975C/7890GC-MS;电子分析天平,箱式电阻炉,涡旋混合器;固相萃取装置;氮吹仪;安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)。2试剂:硫丹、甲氰菊酯、噻嗪酮混合标准溶液;无水硫酸钠、色谱纯乙腈、甲苯;茶叶样品硫丹、甲氰菊酯、噻嗪酮混合标准使用液的配制:(α-硫丹、β-硫丹,硫丹硫酸盐、甲氰菊酯、噻嗪酮)0、0.2、0.4、0.6、0.8、1.0ug/mL。http://ng1.17img.cn/bbsfiles/images/2014/12/201412270910_529372_2266096_3.png3、样品准备将1g粉碎茶叶加到50mL离心试管中,加入3mL水润湿,浸泡10min,加15mL色谱纯乙腈,涡旋2min,静置,过滤,滤液待用,再用15mL色谱纯乙腈重复提取一次,过滤,合并滤液,滤液用50度的水浴中氮吹浓缩至2mL左右待净化。净化 柱子上端装入约1cm高的于650度灼烧过的无水硫酸钠层,以吸附除去多余的水分。a 活化: 4mL 乙腈:甲苯=3:1活化,流出液弃去;b 上样:将待净化的样液加入小柱(安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)),收集流出液;用6mL 乙腈:甲苯=3:1分两次洗涤,合并流出液。c 重新溶解:于50度的水浴中氮吹浓缩至近干,加2mL农残级的正已烷溶解定容,GC-MS 检测。4、仪器条件色谱柱:HP-5MS; 30m*0.25mm*0.25um ;柱温:初始温度80 ºC,维持1min,以30 ºC/min 升温至200 ºC,并保持1min,再以5 ºC/min 升温至280 ºC,保持3 min;流速:1.0mL/min ;载气:高纯氦气;进样量:1μL;进样方式:不分流;进样口温度:230 ºC;接口温度:[
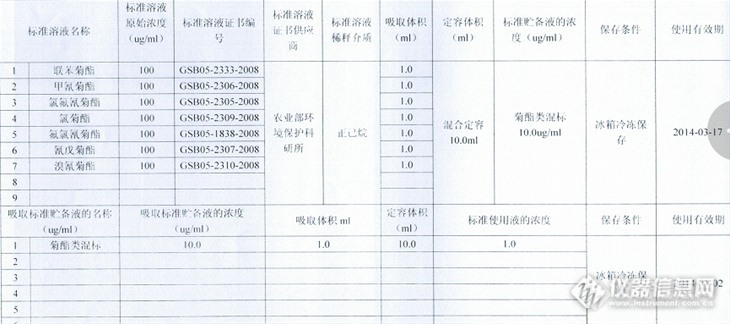
【生活中的仪器分析】食品安全——“菜”米油盐酱醋茶大检测摘要:本文参照GB/T23376-2009、GB/T23204-2008标准,采用安捷伦 气质联用仪Agilent 5975C/7890测定了茶叶中的联苯菊酯、甲氰菊酯、氯氟氰菊酯、氯菊酯、氟氯氰菊酯、氰戊菊酯、溴氰菊酯七种菊酯类农药,结果表明所测定的茶叶均未检出这七种农药残留,是安全可靠的。关键词:GB/T23376-2009;GB/T23204-2008;气质联用仪;七种菊酯类农药;仪器:气质联用仪 Agilent 5975C/7890; 天平: AL204-IC;试剂:联苯菊酯、甲氰菊酯、氯氟氰菊酯、氯菊酯、氟氯氰菊酯、氰戊菊酯、溴氰菊酯七种农药标准溶液(农业部环境保护科研所提供);配制成所需的浓度(见下表)。http://ng1.17img.cn/bbsfiles/images/2013/12/201312221352_483939_2166779_3.png样品前处理简述:称样 5 g于 50 ml试管中,加入水润湿,浸泡 10 min,用15ml乙腈,4200 r/min离心 5 min,离心后收集上层有机相,残渣再用15 ml乙腈提取1次,合并上层有机相,40 ℃水浴旋转蒸发至 2 ml左右,待净化。净化:在活性炭固相萃取柱上端装入2cm高无水硫酸钠,用 10 ml乙腈-甲苯预淋洗小柱后,加入上层有机相,再用 25 ml乙腈-甲苯洗脱,收集全部流出液,浓缩定容至 2.0 ml后进行GCMS分析。气质联用仪操作条件:http://ng1.17img.cn/bbsfiles/images/2013/12/201312221335_483930_2166779_3.png七种菊酯类农药的标准TIC图谱:http://ng1.17img.cn/bbsfiles/images/2013/12/201312221343_483936_2166779_3.png样品两次的平行测定结果的TIC图谱:http://ng1.17img.cn/bbsfiles/images/2013/12/201312221346_483937_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/12/201312221346_483938_2166779_3.png结论:样品与七种菊酯类农药残留的TIC图谱比较可知:所检样品茶叶均未检出这七种菊酯类农药,是安全可靠的,另外这七种菊酯类混标在上述色谱条件下分离效果良好。
按照GB 23200.121-2021方法做农残,改用乙腈,0.2ml/min的流速,做为流动相提取。比例是97:3的甲酸铵:乙腈。氰戊菊酯、氟氰戊菊酯等菊酯类和阿维菌素的相应很低,浓度50ng/ml的单标去跑,相应也才3次方。不知道大家有没有遇到这样的问题?改用乙腈去跑,是因为乙腈跑出来的峰高又窄比用甲醇跑出来的其他物质会好看,做样品时影响更小。不知道哪位大神也用过乙腈去跑121的。请指教下。
我们实验室刚刚成立准备测以下几种:1、甲氰菊酯 2、氯氰菊酯 3、联苯菊酯 4、氯氟氰菊酯 5氟胺氰菊酯 6氟氯氰菊酯 7三唑酮 8、百菌清 9溴氰菊酯 10氰戊菊酯 由于标样是市里面用去年配的标样给我们使用的 感觉被我拿回来之后由于一开始操作不当,使用不小心可能被污染 一、 在用ECD跑进样后基线老漂 会有翘尾 接近200多差距 二、而且跑出的峰感觉有好多杂峰 怕被污染了 是不是一种菊酯有几个同分异构体就有几个峰啊? 三、请教做过农残有机氯和菊酯的哥哥姐姐们给个参数设置 或者提供个方法设置,万分感激 柱子是 HP-5
有参加ACAS-PT243(2016)果蔬汁中毒死蜱、氯氰菊酯、多效唑、哒螨灵农药残留能力验证的吗?加群交流一下啊595456862
各位大虾:哪位可以提供《SN/T1117-2002,2003进出口茶叶中多种菊酯类农药残留检验方法》,《GDFB214-2003茶叶中硫丹、拟除虫菊酯的农残检验方法》。这些是商检局的检验方法。谢谢!
新手刚接触,求问各位大佬像氟氯氰菊酯这种有多个峰的农残积分要合成一个吗?信噪比是看三个峰各自的还是怎么弄?761标准上的检出限是总的检出限还是各自分开的意思?
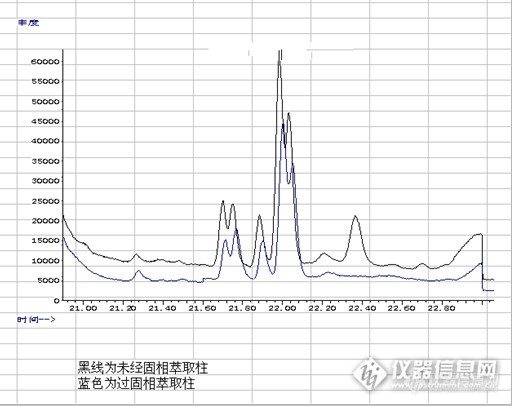
茶叶中菊酯类农药残留的检测仪器:安捷伦5975C/7890GC-MS;电子分析天平,箱式电阻炉,涡旋混合器;固相萃取装置;氮吹仪;Welchrom GraphiCarb/NH2 固相萃取小柱(WSGN010605;500mg/6mL)。试剂:菊酯类混合标准溶液:氯化钠、无水硫酸钠、正已烷、丙酮均为色谱纯;茶叶样品菊酯类混合标准使用液的配制:(联苯菊酯,顺式氰戊菊酯、溴氰菊酯、氰戊菊酯、氯氰菊酯、氯菊酯、氟氰戊菊酯)0、1、2、3ug/mL。http://ng1.17img.cn/bbsfiles/images/2014/12/201412201537_528053_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412201537_528054_2266096_3.png样品准备(1)将1g粉碎茶叶加到50mL离心试管中,加入3mL水润湿,加1g氯化钠,加10mL(1+1)正已烷:涡旋2min,静置,过滤,滤液待用,再用10mL(1+1)正已烷重复提取一次,合并滤液,滤液用50度的水浴中氮吹浓缩至近干,加3mL正已烷溶解残渣作为样液待净化。净化 柱子上端装入约1cm高的于650度灼烧过的无水硫酸钠层,以吸附除去多余的水分。a 活化: 4mL 正己烷:丙酮=9:1活化,流出液弃去;b 上样: 将待净化的样液加入小柱(WSGN010605;500mg/6mL),收集流出液;用6mL的正己烷:丙酮=9:1分两次洗涤,合并流出液。c 重新溶解:于50度的水浴中氮吹浓缩至近干,加2mL农残级的正已烷溶解定容,GC-MS 检测。4、色谱条件色谱柱:货号:WM-5MS 30m*0.25mm*0.25um ;Wel 00215-31043 Ser NO: 661201518 柱温:初始温度80 ºC,维持1min,以10 ºC/min 升温至200 ºC,并保持2min,再以5 ºC/min 升温至230 ºC,保持5 分钟;再以5 ºC/min 升温至280 ºC,保持5 分钟。流速:1.0mL/min ;进样量:1μL; 进样方式:不分流;进样口温度:230 ºC;接口温度:280 ºC ;离子源温度:230 ºC;扫描方式:选择离子扫描(SIM);离子源:EI源,70eV。混标3ug/mL七种菊酯类农药混标的TIC图:ht







 我要推广仪器
我要推广仪器
 下载APP
下载APP