高分子表征技术专题——荧光关联光谱在高分子单链研究中的应用
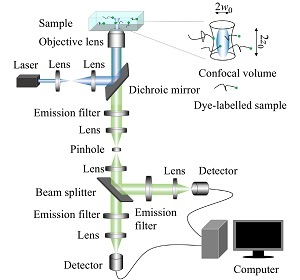
2021年,《高分子学报》邀请到国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。高分子表征技术专题前言孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意! 原文链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304.2020.20238《高分子学报》高分子表征技术专题链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304荧光关联光谱在高分子单链研究中的应用周超 1,2 ,杨京法 1,2 ,赵江 1,2 1.中国科学院化学研究所机构 北京 1001902.中国科学院大学机构 北京 100049作者简介: 赵江,男,1967年生. 分别于1989年、1992年在吉林大学物理系获得学士、硕士学位,1995年于中国科学院物理研究所获得博士学位,之后分别于北京大学化学与分子工程学院、日本产业综合研究所、美国伊利诺伊大学从事博士后研究,2004年起于中国科学院化学研究所任研究员,入选中国科学院“百人计划”,2009年获得国家杰出青年科学基金资助,2013年当选美国物理学会Fellow. 以单分子荧光显微与光谱方法开展关于高分子物理基础性研究,研究方向包括:多电荷大分子、聚合物表界面、高分子动力学、相变与玻璃化转变等 通讯作者: 赵江, E-mail: jzhao@iccas.ac.cn摘要: 荧光关联光谱(fluorescence correlation spectroscopy,FCS)是一项用于研究体系动力学性质的统计光谱技术,随着它被引入材料与化学研究领域,近年来取得了大量全新的研究成果. 该技术在高分子科学研究中也逐渐发挥出越来越大的作用,特别是在聚合物结构和动力学方面,这表明它在高分子领域的巨大潜力. 本文将从FCS的基本原理、实验技巧以及在一些具有挑战性体系中的应用等方面展开,着重介绍它在高分子溶液,如聚电解质溶液、高分子混致不溶现象,以及不同的表界面体系中取得的新成果,展示FCS区别于其他传统技术的特点和优势.关键词: 荧光关联光谱 / 高分子 / 聚电解质 / 表界面 / 混致不溶 目录1. 荧光关联光谱的基本原理2. 荧光关联光谱的实验技巧2.1 实验样品的标记和纯化2.2 激发体积的校准3. 荧光关联光谱在高分子单链研究中的应用3.1 FCS在聚电解质体系中的应用3.2 FCS在高分子混致不溶现象中的应用3.3 FCS在表界面体系中的应用3.4 FCS在有外场作用的体系中的应用4. 荧光关联光谱技术的发展和应用5. 结论参考文献高分子物理研究的目标之一是探究聚合物在不同尺度上的结构与动力学,及其对于高分子体系性质的决定性. 其中,聚合物构象是最为基础的研究内容. 高分子构象是指由于主链上单键内旋转而产生的分子链在空间的不同形态. 对于中性聚合物体系,由于分子链的结构自相似性,利用标度理论可以成功描述其在良溶剂、θ溶剂以及不良溶剂中分子链的尺寸. 散射技术是研究高分子链构象最成功的方法,如:光散射、X射线散射以及中子散射. 就动态光散射而言,它通过检测高分子溶液散射光强随时间涨落而得到其关联函数,从而获得单分子链的扩散速率信息,并获得分子链的流体力学半径信息[1,2]. 结合静态散射实验所获得的回转半径,可以确定聚合物在溶液中的形态[3,4]. 虽然光散射方法在具有短程相互作用的中性聚合物体系表征中非常成功,但是该项技术在一些条件或情形下却遇到了很大的困难,如:多电荷体系、多组分复合体系、表界面体系等. 在多电荷体系中,多重长程静电相互作用使得动态光散射信号中出现令人费解的“快慢模式”[5~7]. 用光散射法来考察高分子的混致不溶现象时,混合溶液中强烈的组分涨落导致强烈的光散射背景信号,严重影响了光散射对信息的提取[8]. 因此,采用新的技术和研究方法开展高分子表征无疑是重要的.荧光关联光谱(fluorescence correlation spectroscopy,FCS)是表征高分子的有效新方法之一. 它与动态光散射同属于光子相关光谱技术,通过分析光信号的涨落而得到分子链动力学信息. 然而,FCS具有很高的探测灵敏度,通过获取荧光涨落信号而得到单个分子的动力学信息. 荧光关联光谱技术是由Madge、Elson和Webb[9~11]在20世纪70年代发展起来的,20世纪90年代,随着Rigler等[12]将共聚焦技术引入,FCS得到快速发展. 采用共聚焦显微技术,FCS的激发-探测空间体积缩小至~10−15 L,激发-探测空间内的分子数目大大地降低,实验的信噪比也随之提高. 与此同时,具有很高灵敏度的单光子检测器的采用使得FCS实现了单分子水平的测量. 随着计算机技术的进步,数据采集卡能够实时地进行数据的采集和相关性计算,使得FCS技术得到了重要的突破,在科学研究中的应用也越来越广泛.近年来,FCS在高分子物理研究中逐渐表现出重要作用,相比于传统的散射技术,它有着独特的优势. 第一,FCS具有极高的灵敏度,可以在极稀薄条件下(~10−9 molL−1)进行测量,同时具有达到光学衍射极限空间分辨率(~200 nm)与出色的时间分辨率(10−6 s). 第二,FCS的信噪比与聚合物的分子量无关. 在实验中,聚合物链通过化学键合的方式实现一比一的荧光标记,因此,分子量不同的样品对于信号的贡献相同. 但是,对于光散射技术而言,散射光强与聚合物分子量具有依赖性,因而信噪比也随之改变,分子量偏小样品的实验难度较大. 第三,对样品的荧光标记同样带来了可选择性与识别性,实现了同一体系中不同组分的区分式研究. 例如,通过对不同组分使用不同的荧光分子进行标记,采用多色FCS对各组分间的运动及其关联进行分析;也可选择性地对多组分体系中的特定组分进行标记,实现复杂体系中特定组分的研究.伴随着FCS技术的发展以及与其他研究手段的联用,其应用越来越广泛,从最初的生物领域[13~15]到胶体[16,17]、聚合物[18,19],从溶液[20~23]到熔体[24~26]、凝胶[27~29]、表界面体系[30~32]等,都取得了许多原创性的成果. 值得指出的是,FCS在测量平动和转动扩散系数、反应速率常数、平衡结合常数、细胞内粒子浓度等方面有着突出的优势[33~35].1. 荧光关联光谱的基本原理当一个体系处于热力学平衡态时,分子的热运动会导致体系浓度、密度等发生局部涨落. 通过相关分析方法,计算这些局部涨落的关联函数,就可以从信号中提取出体系的热力学信息. 动态光散射技术正是运用了此方法,通过测量溶液的散射光强随时间涨落而获得其关联函数,从而获得样品的动力学信息. 荧光关联光谱测量共聚焦空间内样品荧光强度随时间的涨落,通过计算其关联函数而得到对涨落有贡献的热力学性质信息.在激发空间内在任一时刻荧光强度F(t),激发空间内荧光信号在t时刻的强度涨落δF(t)为:其中,⟨F(t)⟩=1/T∫0TF(t)dt,为从0到T 时间内的平均荧光强度.上述涨落的归一化自关联函数为G(τ):自关联函数包含了导致共聚焦空间内荧光信号强度涨落的所有信息,如:平动及转动扩散导致的荧光信号涨落、探针的光物理和化学变化(如:三重态)等导致的涨落等. 对于单光子激发体系,激发空间内的光强分布满足三维高斯分布,对在溶液中进行三维扩散的荧光分子而言,其浓度的涨落满足扩散方程,因而其关联函数的表达式为:其中,Veff=π1.5w02z0为激发空间的体积,特征时间τD=w02/4D为荧光分子通过激发空间所需的平均时间. G(0)=1/Veff⟨c⟩=1/N为激发空间内荧光分子平均数目的倒数,当样品的浓度越低时,G(0)值越大.从G(τ)的表达式可知,FCS的自关联函数有4个变量w0、z0、⟨c⟩、D,其中w0、z0属于仪器的参数,即共聚焦空间的横向半径与纵向半高度,而⟨c⟩、D分别是荧光分子的平均浓度和扩散系数. 因此,在准确标定仪器参数w0w0、z0z0的条件下,通过数值拟合将得到未知样品的浓度和扩散系数. 扩散分子的流体力学半径可以根据Stokes-Einstein方程得到:其中,kB为玻尔兹曼常数,T为温度,η为介质黏度.FCS仪器结构如图1所示,激光器的输出光经过准直扩束后由二向色镜反射进入物镜,并经物镜聚焦在样品中激发荧光. 产生的荧光由同一物镜收集,再次通过二向色镜以及滤镜将杂散的激光以及背景光过滤压制,最终由透镜聚焦并由针孔进行空间滤波进入到检测收集系统.图 1Figure 1. Schematic illustration of instrument structure of fluorescence correlation spectroscopy.由于单光子检测器可能出现接收一个光子产生多个电子的情况,为了消除这个过程带来的误差,可以将荧光信号分成等强度的两部分,然后对2个通道内的信号作交叉关联:2. 荧光关联光谱的实验技巧由于一般的聚合物不发光,因此FCS实验所采用的样品需要进行荧光标记. 另外,在实验操作方面,最需要注意对于激发体积的严格校准,以确保实验测量的准确性.2.1 实验样品的标记和纯化样品标记方法主要有以下2种:第一,在样品需要标记的位点预留反应的基团,如:氨基、羧基、叠氮基团等,再根据不同的基团及FCS实验的要求选择合适的活性荧光分子进行化学键合. 为了获得较高的标记效率,在标记过程中加入的荧光分子的量远大于聚合物,所以反应结束后有大量游离的自由荧光分子存在,需要通过体积排除色谱和超滤等方法进行分离提纯,直至滤液中不再检测到荧光信号.第二,在样品合成过程中加入适当比例的共聚合荧光单体进行共聚,例如,通过RAFT聚合制备聚异丙基丙烯酰胺(PNIPAM)时,可以加入适当比例的荧光单体来合成具有一定分子量范围、分子量分布较窄和荧光标记的样品[36]. 反应完成后同样也需要超滤、透析等方式进行分离提纯.2.2 激发体积的校准FCS实验之前,需要对仪器进行校正得到仪器激发体积的参数. 采用已知浓度和扩散系数的荧光分子样品来进行校正,例如Rhodamine 6G (Rh6G)分子,它在纯水中的扩散系数为414 μm2s−1 (25 °C),实验中一般将其配置成5×10−9 molL−1 (5 nmolL−1)的水溶液进行FCS测量,然后通过对测得的关联函数进行拟合即可得到激发空间的尺寸.另外,温度对于扩散系数的影响很大,不同温度下进行实验时,同样需要对扩散系数进行校正,校正的公式如下:如图2所示,以波长为488 nm的激光作为激发光,对FCS测量得到的Rhodamine 6G的自相关曲线进行拟合得到激发空间的尺寸为w0=0.224 μm,z0=1.608 μm.图 2Figure 2. A typical autocorrelation function curve and the fitting result of free Rhodamine 6G molecules in water.需要说明的是,FCS的测量会受到样品体系折射率不匹配的影响. 如图3所示,当样品溶液与物镜的折射率不匹配时,会导致表观的激发体积出现显著变化:第一,表观的w0值随折射率不匹配的增加而减小,这是折射率不匹配产生的像差导致;第二,随着物镜焦点位置从界面处愈加深入到样品溶液中时,折射率不匹配导致的表观w0值的变化愈明显[36].图 3Figure 3. (a) Representative normalized autocorrelation function curves of fluorescent nanoparticles diffusing in aqueous solution of glycerol at a small focal depth (25 μm) (b) Values of the apparent lateral radius of the excitation-detection volume of FCS as a function of the refractive index of the solution. The distance of the focal point in the sample medium away from the coverslip surface is displayed. (Reprinted with permission from Ref.[36] Copyright (2012) American Chemical Society).依据FCS的原理,w20=4DτDw02=4DτD,因此,即使微小w0变化也将显著影响探针分子拟合得到的扩散系数值. 因此,选择合适的溶液体系和物镜使得折射率尽可能匹配,对于FCS的测试准确性至关重要. 在折射率不匹配问题无法避免时,如图3(b)中,可以使用一个较低的焦点位置(25 μm)能有效地避免激发体积的畸变[36].此外,如图4所示,以厚度为0.16 mm的盖玻片为例,当实验使用物镜的校正环与样品池底部的盖玻片厚度不匹配时,激发体积的尺寸也会出现较大的偏差,所以在实验前还需注意物镜校正环与盖玻片厚度是否匹配[37].图 4Figure 4. Values of the apparent lateral radius of the excitation-detection volume of FCS as a function of the value of correcting collar (Reprinted with permission from Ref.[37] Copyright (2018) University of Chinese Academy of Sciences).因此,在FCS实验中,应该尽量选择合适的物镜类型以匹配样品的折射率,并调整镜头校正环数值与盖玻片厚度一致,如果折射率不匹配的情况不能避免,那就选择较低的、固定的焦点深度值以保证实验结果可靠可信.除了上述两点之外,在实验过程中还需要注意激光光强的选择,过强的入射光容易导致荧光探针发生光漂白而带来实验误差,因此应该降低进入物镜的激光光强进行实验.3. 荧光关联光谱在高分子单链研究中的应用FCS以其独特的优势在一些传统研究手段难以涉足的高分子体系中展现出独特的优势,例如:考察水溶液中聚电解质的单链动力学[38~44]、混致不溶现象中高分子链构象的变化[36]、表界面体系中高分子的扩散动力学[30~32,45~48]等等.3.1 FCS在聚电解质体系中的应用聚电解质是主链或者侧链上带有可离子化基团的聚合物,在极性溶剂中,聚电解质主链由于解离而带电,同时存在大量带有相反电荷的抗衡离子[49,50]. 正是聚电解质链间、链段间以及链与抗衡离子间多重长程静电相互作用,在赋予聚电解质丰富性质的同时,也给聚电解质的研究带来了很大的困难[51~53]. 例如,当采用动态光散射技术研究带电聚合物体系时,在低离子强度的聚电解质溶液中,存在“快与慢”的2种松弛模式. 为了探究聚电解质中的这种多级松弛模式的起源,研究人员进行了大量的实验并提出了多种可能的解释,但至今仍未有一个确切的回答[5,6,54~56].如果采用传统散射技术来研究低离子强度条件下带电聚合物体系的扩散运动,实验中遇到不少困难,而FCS实验中样品极稀浓度和极高选择性的优势就体现出来,依靠FCS技术,研究人员可以在极稀薄条件下进行实验研究,在聚电解质溶液体系获得全新的信息.Wang等[38]利用FCS在实验上第一次观察到了在无扰溶液中疏水聚电解质的一级构象转变. 如图5(a)所示,弱聚电解质聚(2-乙烯基吡啶) (P2VP)分子的构象随带电分数的变化而呈现出一级转变特征,即:随pH的升高由伸展的线团构象至坍缩的链球. 除了通过pH值改变聚电解质的带电分数,聚电解质的构象转变也可以由改变外加盐的浓度导致,即:抗衡离子吸附与静电屏蔽作用. 如图5(b)所示,P2VP的单分子链流体力学半径随着静电屏蔽长度的增加而连续增加.图 5Figure 5. (a) Diffusion coefficient of P2VP as a function of pH value of the solution. Inset: The hydrodynamic radius of P2VP as a function of pH value (b) The hydrodynamic radius of P2VP as a function of Debye length of the system (Reprinted with permission from Ref.[38] Copyright (2007) American Institute of Physics).Xu等[39]利用FCS技术在单分子水平上研究了强聚电解质的构象. 实验发现,在无外加盐的情况下,强聚电解质聚苯乙烯磺酸钠(NaPSS)和季胺化聚(4-乙烯基吡啶)(QP4VP)的流体力学半径和聚合度之间分别存在着0.7和0.9的标度关系,说明在低离子强度时,聚电解质链的构象比中性聚合物在良溶剂中溶胀的无规线团构象更加伸展. 如图6所示,采用棒状构象的分子模型得到了理想的拟合结果(其中QP4VP在高分子量部分出现偏离是高分子量聚电解质吸附更多的抗衡离子所导致的). 拟合结果显示分子链的直径分别为2.2和2.3 nm,这比理论假设的裸露水合聚电解链的直径0.8 nm要大很多,这也说明了聚电解质链的周围有抗衡离子云的存在.图 6Figure 6. Values of hydrodynamic radius of NaPSS and QP4VP plotted as a function of degree of polymerization. The solid lines denote the numerical fitting based on the theoretical model of diffusion of a rod-like molecule, and the dashed line denotes the fitting results using the diameter of a hydrated chain, i.e., d=0.8 nm. (Reprinted with permission from Ref.[39] Copyright (2016) American Institute of Physics).Xu等[40]进一步研究了在不同外加盐浓度情况下聚电解质链的构象. 如图7所示,聚电解质分子链构象具有分子量依赖性:在低盐浓度时,短链分子的聚电解质采取棒状构象,而长链分子采取无规线团构象;随着外加盐浓度的增加,所有的NaPSS和QP4VP均采取无规线团构象.图 7Figure 7. Diffusion coefficient of NaPSS (a) and QP4VP (b) as a function of degree of polymerization under salt concentrations of 10−4, 0.1, and 1.0 molL−1, respectively The solid lines represent the results of fitting using the relation of Rh∼N−v. (Reprinted with permission from Ref.[40] Copyright (2018) American Institute of Physics).Ren等[41]通过FCS技术研究了i-motif DNA的解折叠过程. 如图8所示,在不同盐浓度的条件下,随着pH值的升高,i-motif DNA均发生了从有序的四联体结构到无规线团的构象转变,并且这一转变对盐浓度有着依赖性:盐浓度越高,解折叠的起始pH值就越低. 这种盐浓度依赖性的主要原因是外加盐的引入导致更多的抗衡离子吸附在DNA链上而降低了链的电荷密度,降低了链周围的局部质子浓度,而后者是控制折叠形成的关键因素.图 8Figure 8. The values of hydrodynamic radius of a single i-motif DNA strand as a function of pH value in the solution Three conditions were chosen: solution without any salt addition (salt-free), and 50 mmolL−1 and 100 mmolL−1 NaCl solutions (physiological environment) The start and end points of the conformation transition are denoted by the arrows. (Reprinted with permission from Ref.[41] Copyright (2018) The Royal Society of Chemistry).如果将光子计数直方图(PCH)技术与FCS相结合,可以对聚电解质主链的电势、有效带电量、抗衡离子分布等方面进行深入研究. 例如,Luo等[42]将pH敏感的荧光探针标记于NaPSS链的不同位点,采用PCH技术测量分子链局部的pH值,发现聚电解质链附近的局部氢离子浓度比本体溶液中高2~3个数量级,而末端效应使得分子链中间的静电势高于末端的静电势. 同时,他们还发现氢离子浓度在径向呈现出e指数衰减的趋势,这证明了聚电解质链周围存在抗衡离子云的说法[43].Jia等[44]研究了抗衡离子分布与聚合物浓度的依赖关系,通过FCS测量NaPSS溶液中作为抗衡离子探针的带负电荧光分子的扩散系数,确定自由探针和吸附于主链的探针2个组分,发现与主链结合的抗衡离子组分随着聚合物浓度的增加而增加. Xu等[40]采用PCH测量NaPSS单分子链电位,发现其随着聚合度的增大而单调上升,且在聚合度大的区间达到饱和. 这说明主链的静电势与分子量不是线性关系,其有效带电分数以及有效电荷密度随着分子量的增加而减小. 上述实验结果说明聚电解质抗衡离子与主链的相互作用是吸附与脱附的动态平衡,而不是经典的Manning抗衡离子凝聚[57~60].3.2 FCS在高分子混致不溶现象中的应用高分子的混致不溶现象(cononsolvency)是一类回归型过程:2种高分子的良溶剂按一定比例混合后反而成为了不良溶剂[61,62]. 一个典型的例子是:常温下聚异丙基丙烯酰胺(PNIPAM)在水与一定比例的甲醇、乙醇、异丙醇、丙酮、四氢呋喃、DMSO等良溶剂的混合液中不再溶解,溶液的相分离温度显著改变,溶液黏度下降,PNIPAM凝胶溶胀率下降. 研究人员对这一现象的起源进行了大量的实验探究,至今未能达成共识[8,63~66].了解高分子链的构象对于理解混致不溶现象至关重要. 前人采用光散射方法研究了水和甲醇混合溶剂中PNIPAM链从线团到塌缩球再到线团的构象转变[64]. 需要特别说明的是,为了在极稀溶液中获得足够高的散射强度与信噪比,研究中采用了分子量高达107 gmol−1的样品. 当采用FCS技术研究该过程时,由于其超高的灵敏度以及与样品分子量无关的信噪比,可在混合溶剂环境下高分子单链的研究中提供独特的信息[67]. Wang等[36]利用FCS研究了PNIPAM在水-乙醇混合溶剂中的混致不溶过程. 如图9所示,PNIPAM具有非对称的回归型构象变化特征:随着乙醇浓度的增大,在一个很窄的乙醇浓度范围内PNIPAM链剧烈塌缩,然后在很宽的乙醇浓度范围内逐渐地再度伸展,说明这一构象转变不是先前文献中所认为的一级构象转变过程. 这表明乙醇分子比水分子更强烈地与PNIPAM链发生作用,这是由乙醇较强的疏水水合效应所致,暗示了Tanaka提出的模型中水合/失水的协同能力强于醇分子吸附/脱附的协同能力[65,66].图 9Figure 9. Normalized autocorrelation function curves of diffusing single chains of PNIPAM with five degrees of polymerizations in pure ethanol (a) and at xEtOHxEtOH of 0.25 (b) The solid line with each data set denotes the results of the numerical fitting using three-dimensional diffusion model Rh6G in (a) denotes the results of free fluorescent Rhodamine 6G, and its drastic difference from those of polymers indicates the successful labeling and sample purification (c) The values of hydrodynamic radius of PNIPAM single chains as a function of xEtOHxEtOH (Reprinted with permission from Ref.[36] Copyright (2012) American Chemical Society).如图10所示,不同乙醇浓度下得到PNIPAM单链的尺寸的标度率(Rh∼NυRh∼Nυ)表明,标度指数νν随着xEtOHxEtOH变化:随着乙醇的浓度的增加,ν从~0.57到0.5再到~1/3变化,说明在上述3个区域,PNIPAM高分子链分别采取了溶胀、无规线团、坍缩链球的构象,即:由纯水中的溶胀线团经无规线团构象而急剧转变为塌缩链球构象,进而又再度逐渐伸展,经过无规线团构象变化至溶胀线团构象. 从标度指数的变化也可以发现回归型链构象变化的高度非对称性,进一步印证了Tanaka提出的协同吸附-优先吸附模型[65,66].图 10Figure 10. Typical double-logarithmic plot of hydrodynamic radius of single PNIPAM chains as a function of degree of polymerization under different solvent compositions: (a)xEtOH=xEtOH=1.0, (b)xEtOH=xEtOH=0.28, (c)xEtOH=xEtOH=0.25 Solid lines are the least-squares linear fitting (d) The vv values as a function of xEtOHxEtOH The three dotted lines denote the theoretical values of the static scaling index for a random coil (0.588), an undisturbed coil (0.5), and a compact globule (1/3). (Reprinted with permission from Ref.[36] Copyright (2012) American Chemical Society).3.3 FCS在表界面体系中的应用受限高分子链,尤其是处于界面的高分子链结构及动力学性质,直接关系到表界面的机械性能、摩擦性能、流变性能等,这些性质与高分子材料在表界面上的应用息息相关,如涂料、润滑剂、胶黏剂等[68~71]. 但是对于高分子链在表界面处的动力学研究存在着不少技术难题,主要原因是表界面动力学带来的浓度涨落被局限于二维或准二维空间,探测难度极大,使得传统的散射方法难以应用. 近年来,得益于单分子技术的迅猛发展,空间和时间分辨能力分别有了显著的优化,极大提高了人们直接“观察”分子或粒子行为的能力,这为我们从分子水平认识聚合物在界面上的动力学性质打下了基础.荧光关联光谱因其极高的灵敏度与显微测量能力被成功地应用于表界面体系的研究中. 对于处于二维自由扩散的分子而言,其自关联函数为:其中,w0是二维FCS观察区域(即激发空间在界面等二维平面投影)的半径,⟨ρ⟩=⟨N⟩/A,即单位面积内荧光探针的平均数量,A是激发空间在界面等二维平面上投影的面积.Sukhishvili等[30]利用FCS研究了荧光染料标记的不同分子量的聚乙二醇(PEO)在固-液界面上的扩散. 从分子链界面扩散运动行为出发,分析出在极稀浓度的条件下聚合物分子在固-液界面上呈现出了紧密吸附的pancake构象,发现了界面扩散系数与分子量的-3/2的独特标度率. Zhao等[31,32]则利用FCS研究了PEO在固-液界面上扩散速率与界面吸附浓度的非线性关联性,即:随着聚合物浓度的增加,其扩散系数先增加并在某一浓度值达到极值,进而骤然大幅下降. 这是由于极低浓度分子链紧密吸附的pancake构象会随着吸附浓度的增加变成loop-tail-train构象,即:吸附使得分子链构象变得相对松散,其扩散速率由与基底接触的train部分占主导. 随着吸附浓度的增加,较为自由的loop-tail部分则增加了其运动能力,因此扩散系数增加;更高浓度时扩散系数出现骤降是因为体系中出现了jamming效应,即分子链间的作用增强,阻碍了分子链的扩散运动.Ye等[45]利用FCS研究了不同拓扑结构的聚合物链在石英-二氯甲烷界面上的扩散,如图11所示,线形聚苯乙烯(PS)扩散的标度率为D∼M−1.5,重现了reptation模型;而环形PS的标度率则为D∼M−1,展现为Rouse模型. 两者的差异是由于环形分子没有末端,无法像线形分子一样完成蛇行运动,而是由一系列链段受到热激发进行跳跃,跨过局部能垒的运动组成.图 11Figure 11. Double-logarithmic plots of center-of-mass diffusion coefficient against molecular weight for surface diffusion of cyclic (c-PS) and linear (l-PS) polystyrene chains on fused silica-DCM interface The solid lines with slopes of 1 and 3/2 are drawn as guides to the eye The dashed lines through the points representing the best fit of the data give power law slopes of 1.46 for linear chains and 1.00 for cyclic chains. (Reprinted with permission from Ref.[45] Copyright (2016) The Royal Society of Chemistry)Yang等[46]利用FCS研究了不同盐溶液作为液相时,NaPSS在疏水单层分子膜界面上的扩散行为. 如图12所示,吸附在疏水表面的聚电解质分子链的扩散受到液相中不同阴离子的影响,主要原因在于不同的阴离子效应改变了界面疏水相互作用强度,从而改变了界面与分子链之间摩擦力,造成扩散系数的显著改变.图 12Figure 12. Typical data of the lateral diffusion coefficient of a NaPSS single chain at the interface of a hydrophobic surface and an aqueous solution as a function of the salt concentration in the aqueous solution (Reprinted with permission from Ref.[46] Copyright (2011) American Chemical Society)Yang等[47]利用FCS技术研究了聚苯乙烯与聚异戊二烯(PI)的嵌段共聚物在二甲基甲酰胺(DMF)与PI聚合物构成的液体界面上的扩散运动. 如图13所示,在本体聚合物分子量跨越了2个数量级的变化,界面上PS-b-PI的扩散系数仅有轻微的下降. 这表明,在PI/DMF的体系中,存在很低黏度的界面层,该界面层的黏度与构成界面的本体聚合物的分子量不存在明显依赖性.图 13Figure 13. Interfacial diffusion coefficient of single PS-b-PI chain as a function of the molecular weight of bulk PI The dashed line is for the guide of eye Inset: illustration of the sample geometry (Reprinted with permission from Ref.[47] Copyright (2008) American Chemical Society).Li等[48]利用FCS探究了PEO分子在烷烃-水界面上的扩散行为. 研究发现,PEO在该界面上聚合物的横向扩散为正常扩散,与二维布朗运动模型相吻合. 如图14所示,液-液界面上的PEO的界面扩散系数与其聚合度之间存在D∼N−0.5的标度关系,这一新的标度关系表明其界面扩散运动遵循着新的运动机理.图 14Figure 14. The logarithm of interfacial diffusion coefficient of PEO as a function of the logarithm of molecular weight (Reprinted with permission from Ref.[48] Copyright (2020) The Royal Society of Chemistry).从单分子层面上研究界面扩散,有助于发现分子最真实和原始的扩散行为规律,这在传统的系综平均实验中往往会被忽略或者被多种因素耦合而产生的运动行为掩盖,这是上述FCS实验结果最大的优势之处. 此外,值得注意的是,在研究固-液界面上聚合物扩散机理时,不同研究团队利用FCS和单粒子追踪(single particle tracking, SPT)技术,得到了不同的结果及界面扩散机理,也因此导致了FCS和SPT 2种技术在界面分子动力学研究上存在多年的学术争论[30,31,72,73]. 我们基于这个问题也展开了实验对比,发现FCS和SPT都能够提供准确且可靠的实验结果,在条件满足时两者能够得到相互吻合相互匹配的实验结果,相关数据结果将在未来进行发表.3.4 FCS在有外场作用的体系中的应用对于聚合物而言,在其合成、分离、加工等过程中有可能会经历电场、流动场、剪切场等作用,尤其在生命体中更是常见. 因此,对于外场作用下的聚合物性质的研究也是极为重要的.当我们将荧光关联光谱应用于外场作用下的体系中时,除了分子热运动导致平动扩散引起的荧光信号涨落,还不得不考虑外场导致荧光分子定向运动通过激发体积带来的信号涨落. 带有定向运动的FCS,如果其运动的方向垂直于激光光束的方向,经过修正的模型拟合关联函数可以获得扩散系数与定向运动速率:其中,vf=w0/τf即为定向运动速率.Dong等[74]将FCS和毛细管电泳结合起来测定了量子点在极稀溶液中的表面电势. 利用FCS的自关联函数拟合得到荧光粒子的定向运动速度和扩散系数,在电泳实验中定向运动的特征时间τf和自扩散系特征时间τD之间满足:其中,Q为带电量,E为外加电场强度. 通过测定不同电场强度下定向运动和扩散的特征时间,通过线性拟合得到荧光粒子的表面电势. Wang等[75]利用FCS研究了P2VP在交变电场下的单链构象转变. 结果表明电场强度对于分子链构象的影响存在滞后转变. 这种滞后现象可以归因于单个疏水性聚电解质链的不对称双稳态能态,由于抗衡离子的解离、迁移和凝聚,其coil和globule构象之间的势垒可以通过交变电场诱导的偶极子降低到kBT以下.4. 荧光关联光谱技术的发展和应用随着FCS技术的发展,出现了双色荧光关联光谱(DC-FCCS)[76,77]、双焦点荧光关联光谱[78,79]、FCS与荧光共振能量转移(FRET)联用[80,81]、可连续改变共焦体积荧光关联光谱[82]等新技术. 这些新技术相较于传统的FCS,可以获取样品更多的热力学信息. 图15是DC-FCCS的简单示意图,采用2种波长的激光分别激发2种对应的荧光分子,然后选择性光学器件对不同波长的荧光进行分离,最后由2个APD检测器分别检测2种荧光信号,再对信号进行关联性分析. DC-FCCS的基本原理就不在此赘述,除了对2种荧光分子的荧光强度涨落进行各自的自关联分析之外,我们还可以对这2种荧光信号做交叉关联分析得到两者相互运动乃至相互作用的信息. 需要说明的是,选择的这2种荧光分子在光谱上必须分离得很好,否则会出现很大的串扰影响实验结果.图 15Figure 15. Schematic illustration of dual color fluorescence cross-correlation spectroscopyChen等[83]利用DC-FCCS和光散射相结合的方法深入研究了聚电解质溶液中单链运动之间的关联性,发现了聚电解质分子链间的运动耦合. 将DC-FCCS实验得到自关联函数的自由扩散部分转化为均方位移数据(MSD),发现其在长短2个时间尺度上分别存在具有不同扩散系数的正常扩散运动,表明链间的静电排斥相互作用带来的“笼子效应”导致了单个分子链的自扩散运动中同样存在一快一慢2种时间尺度上的扩散模式:短时间尺度上为“笼子”内的快扩散行为,长时间尺度上为跨越不同“笼子”的慢扩散行为(如图16所示). 这2种松弛模式均存在强烈的离子强度依赖性,随着外加盐浓度的增加,削弱了链间的排斥作用而弱化了“笼子效应”,导致了长短时间尺度上的动力学非均匀性减弱,甚至消失. 实验结果还表明,聚合物浓度的增加限制了聚电解质链的运动,从而削弱了链间运动的关联性(如图16(b)所示). 将其与光散射中“慢模式”对应的扩散系数对比发现,“慢模式”对应的扩散系数数值处于分子链自扩散长短时间尺度的扩散系数之间,这说明光散射观察到的“快慢模式”与长程静电相互作用引起“笼子效应”有着密切的联系,同时也说明聚电解质的多级松弛过程比我们预想的更加复杂.图 16Figure 16. (a) Values of the diffusion coefficient of the short-time diffusion (Dshort-timeDshort-time) and the long-time diffusion (Dlong-timeDlong-time) of NaPSS with three different molecular weights under different salt concentrations (b) Diffusion coefficient of single NaPSS chain with three different molecular weights at short- and long-time lag as a function of concentration Diffusion coefficients measured by DLS (the slow mode, DDLS,slowDDLS,slow) are displayed for comparison. (Reprinted with permission from Ref.[83] Copyright (2019) American Chemical Society).5. 结论荧光关联光谱技术作为一种高灵敏度的显微统计光谱方法,能够有效地在多种复杂条件下开展高分子动力学的研究,包括:极稀薄溶液、表界面等等. 这项技术出色的空间分辨能力以及由于荧光标记带来的分子识别性,赋予了更加丰富的应用能力与前景. 随着这项技术的不断发展和应用范围的进一步拓展,相信未来它会和传统的散射技术一样被越来越多的人了解和使用,在多个领域都能取得丰富且具创造性的成果.致 谢 感谢研究生及合作者的辛勤劳动与贡献.参考文献[1]Wu C, Zhou S. Phys Rev Lett, 1996, 77(14): 3053−3055 doi: 10.1103/PhysRevLett.77.3053[2]Gao J, Wu C. Macromolecules, 1997, 30(22): 6873−6876 doi: 10.1021/ma9703517[3]Liu X B, Luo S K, Ye J, Wu C. Macromolecules, 2012, 45(11): 4830−4838 doi: 10.1021/ma300629d[4]Morishima K, Ishiwari F, Matsumura S, Fukushima T, Shibayama M. Macromolecules, 2017, 50(15): 5940−5945 doi: 10.1021/acs.macromol.7b00883[5]Sedlak M, Amis E J. J Chem Phys, 1992, 96(1): 826−834 doi: 10.1063/1.462468[6]Muthukumar M. Macromolecules, 2017, 50(24): 9528−9560 doi: 10.1021/acs.macromol.7b01929[7]Zhou K, Li J, Lu Y, Zhang G, Xie Z, Wu C. Macromolecules, 2009, 42(18): 7146−7154 doi: 10.1021/ma900541x[8]Hao J, Cheng H, Butler P, Zhang L, Han C C. J Chem Phys, 2010, 132(15): 154902 doi: 10.1063/1.3381177[9]Magde D, Webb W W, Elson E. Phys Rev Lett, 1972, 29(11): 705−708 doi: 10.1103/PhysRevLett.29.705[10]Elson E L, Magde D. Biopolymers, 1974, 13(1): 1−27 doi: 10.1002/bip.1974.360130102[11]Magde D, Elson E L, Webb W W. Biopolymers, 1974, 13(1): 29−61 doi: 10.1002/bip.1974.360130103[12]Rigler R, Mets U, Widengren J, Kask P. Eur Biophys J Biophy, 1993, 22(3): 169−175[13]Dross N, Spriet C, Zwerger M, Muller G, Waldeck W, Langowski J. PLoS One, 2009, 4(4): e5041 doi: 10.1371/journal.pone.0005041[14]Mtze J, Ohrt T, Schwille P. Laser Photonics Rev, 2011, 5(1): 52−67 doi: 10.1002/lpor.200910041[15]Schwille P, Haupts U, Maiti S, Webb W W. Biophys J, 1999, 77(4): 2251−2265 doi: 10.1016/S0006-3495(99)77065-7[16]Xie J, Nakai K, Ohno S, Butt H J, Koynov K, Yusa S. Macromolecules, 2015, 48(19): 7237−7244 doi: 10.1021/acs.macromol.5b01435[17]Caruso F, Donath E, Mohwald H. J Phys Chem B, 1998, 102(11): 2011−2016 doi: 10.1021/jp980198y[18]Vagias A, Raccis R, Koynov K, Jonas U, Butt H J, Fytas G, Kosovan P, Lenz O, Holm C. Phys Rev Lett, 2013, 111(8): 088301 doi: 10.1103/PhysRevLett.111.088301[19]Lumma D, Keller S, Vilgis T, Radler J O. Phys Rev Lett, 2003, 90(21): 218301 doi: 10.1103/PhysRevLett.90.218301[20]Cherdhirankorn T, Best A, Koynov K, Peneva K, Muellen K, Fytas G. J Phys Chem B, 2009, 113(11): 3355−3359 doi: 10.1021/jp809707y[21]Schaeffel D, Yordanov S, Staff R H, Kreyes A, Zhao Y, Schmidt M, Landfester K, Hofkens J, Butt H J, Crespy D, Koynov K. ACS Macro Lett, 2015, 4(2): 171−176 doi: 10.1021/mz500638e[22]Jee A Y, Cho Y K, Granick S, Tlusty T. P Natl Acad Sci USA, 2018, 115(46): E10812 doi: 10.1073/pnas.1814180115[23]Jee A Y, Dutta S, Cho Y K, Tlusty T, Granick S. P Natl Acad Sci USA, 2018, 115(1): 14−18 doi: 10.1073/pnas.1717844115[24]Cherdhirankorn T, Floudas G, Butt H J, Koynov K. Macromolecules, 2009, 42(22): 9183−9189 doi: 10.1021/ma901439u[25]Cherdhirankorn T, Harmandaris V, Juhari A, Voudouris P, Fytas G, Kremer K, Koynov K. Macromolecules, 2009, 42(13): 4858−4866 doi: 10.1021/ma900605z[26]Doroshenko M, Gonzales M, Best A, Butt H J, Koynov K, Floudas G. Macromol Rapid Commun, 2012, 33(18): 1568−1573 doi: 10.1002/marc.201200322[27]Michelman-Ribeiro A, Boukari H, Nossal R, Horkay F. Macromolecules, 2004, 37(26): 10212−10214 doi: 10.1021/ma048043d[28]Zustiak S P, Boukari H, Leach J B. Soft Matter, 2010, 6(15): 3609−3618 doi: 10.1039/c0sm00111b[29]Modesti G, Zimmermann B, Borsch M, Herrmann A, Saalwachter K. Macromolecules, 2009, 42(13): 4681−4689 doi: 10.1021/ma900614j[30]Sukhishvili S A, Chen Y, Muller J D, Gratton E, Schweizer K S, Granick S. Nature, 2000, 406(6792): 146 doi: 10.1038/35018166[31]Zhao J, Granick S. Macromolecules, 2007, 40(4): 1243−1247 doi: 10.1021/ma062104l[32]Zhao J, Granick S. J Am Chem Soc, 2004, 126(20): 6242−6243 doi: 10.1021/ja0493749[33]Ries J, Schwille P. Bioessays, 2012, 34(5): 361−368 doi: 10.1002/bies.201100111[34]Elson E L. Methods Enzymol, 2013, 518: 1−10 doi: 10.1016/B978-0-12-388422-0.00001-7[35]Papadakis C M, Kosovan P, Richtering W, Woll D. Colloid Polym Sci, 2014, 292(10): 2399−2411 doi: 10.1007/s00396-014-3374-x[36]Wang F, Shi Y, Luo S J, Chen Y M, Zhao J. Macromolecules, 2012, 45(22): 9196−9204 doi: 10.1021/ma301780f[37]Zheng Kaikai(郑锴锴). Dynamics of a Single Polymer Chain under Shear(剪切场下聚合物分子单链动力学行为研究). Doctoral Dissertation of University of Chinese Acdemy of Sciences((中国科学院大学博士学位论文), 2018.[38]Wang S, Zhao J. J Chem Phys, 2007, 126(9): 091104 doi: 10.1063/1.2711804[39]Xu G, Luo S, Yang Q, Yang J, Zhao J. J Chem Phys, 2016, 145(14): 144903 doi: 10.1063/1.4964649[40]Xu G, Yang J, Zhao J. J Chem Phys, 2018, 149(16): 163329 doi: 10.1063/1.5035458[41]Ren W, Zheng K, Liao C, Yang J, Zhao J. Phys Chem Chem Phys, 2018, 20(2): 916−924 doi: 10.1039/C7CP06235D[42]Luo S J, Jiang X B, Zou L, Wang F, Yang J F, Chen Y M, Zhao J. Macromolecules, 2013, 46(8): 3132−3136 doi: 10.1021/ma302276b[43]Luo Shuangjiang(罗双江), Gao Peiyuan(高培源), Guo Hongxia(郭洪霞), Yang Jingfa(杨京法), Zhao Jiang(赵江). Acta Polymerica Sinica(高分子学报), 2017, (9): 1479−1487 doi: 10.11777/j.issn1000-3304.2017.17065[44]Jia P, Yang Q, Gong Y, Zhao J. J Chem Phys, 2012, 136(8): 084904 doi: 10.1063/1.3688082[45]Ye S, Tang Q, Yang J, Zhang K, Zhao J. Soft Matter, 2016, 12(47): 9520−9526 doi: 10.1039/C6SM02103D[46]Yang Q, Zhao J. Langmuir, 2011, 27(19): 11757−11760 doi: 10.1021/la202510d[47]Yang J F, Zhao J, Han C C. Macromolecules, 2008, 41(20): 7284−7286 doi: 10.1021/ma8015135[48]Li Z, Yang J F, Hollingsworth J V, Zhao J. RSC Adv, 2020, 10(28): 16565−16569 doi: 10.1039/D0RA02630A[49]Oosawa F. Polyelectrolytes. New York: Marcel Dekker, 1971[50]Dobrynin A V, Rubinstein M. Prog Polym Sci, 2005, 30(11): 1049−1118 doi: 10.1016/j.progpolymsci.2005.07.006[51]Forster S, Schmidt M, Antonietti M. Polymer, 1990, 31(5): 781−792 doi: 10.1016/0032-3861(90)90036-X[52]Fuoss R M. J Polym Sci, 1948, 3(4): 603−604 doi: 10.1002/pol.1948.120030414[53]Muthukumar M. J Chem Phys, 2004, 120(19): 9343−9350 doi: 10.1063/1.1701839[54]Mattoussi H, Karasz F E, Langley K H. J Chem Phys, 1990, 93(5): 3593−3603 doi: 10.1063/1.458791[55]Reed W F, Ghosh S, Medjahdi G, Francois J. Macromolecules, 1991, 24(23): 6189−6198 doi: 10.1021/ma00023a021[56]Li J, Li W, Huo H, Luo S, Wu C. Macromolecules, 2008, 41(3): 901−911 doi: 10.1021/ma071284b[57]Manning G S. J Chem Phys, 1969, 51(3): 924−933 doi: 10.1063/1.1672157[58]Manning G S. J Chem Phys, 1969, 51(3): 934−938 doi: 10.1063/1.1672158[59]Manning G S. J Chem Phys, 1969, 51(8): 3249−3252 doi: 10.1063/1.1672502[60]Manning G S. Biophys Chem, 1977, 7(2): 95−102 doi: 10.1016/0301-4622(77)80002-1[61]Schild H G, Muthukumar M, Tirrell D A. Macromolecules, 1991, 24(4): 948−952 doi: 10.1021/ma00004a022[62]Winnik F M, Ringsdorf H, Venzmer J. Macromolecules, 1990, 23(8): 2415−2416 doi: 10.1021/ma00210a048[63]Chee C K, Hunt B J, Rimmer S, Soutar I, Swanson L. Soft Matter, 2011, 7(3): 1176−1184 doi: 10.1039/C0SM00836B[64]Zhang G Z, Wu C. J Am Chem Soc, 2001, 123(7): 1376−1380 doi: 10.1021/ja003889s[65]Tanaka F, Koga T, Kojima H, Xue N, Winnik F M. Macromolecules, 2011, 44(8): 2978−2989 doi: 10.1021/ma102695n[66]Kojima H, Tanaka F. Soft Matter, 2012, 8(10): 3010−3020 doi: 10.1039/c2sm06883d[67]Grabowski C A, Mukhopadhyay A. Phys Rev Lett, 2007, 98(20): 207801 doi: 10.1103/PhysRevLett.98.207801[68]Fleer G J. Adv Colloid Interface Sci, 2010, 159(2): 99−116 doi: 10.1016/j.cis.2010.04.004[69]Granick S, Bae S C. J Polym Sci, Part B: Polym Phys, 2006, 44(24): 3434−3435 doi: 10.1002/polb.21004[70]Granick S, Kumar S K, Amis E J, Antonietti M, Balazs A C, Chakraborty A K, Grest G S, Hwaker C J, Janmey P, Kramer E J, Nuzzo R, Russell T P, Safinya C R. J Polym Sci, Part B: Polym Phys, 2003, 41(22): 2755−2793 doi: 10.1002/polb.10669[71]Guo Z Y, Cao X L, Guo L L, Zhao Z Y, Ma B D, Zhang L, Zhang L, Zhao S. J Dispersion Sci Technol, 2020, Doi:10.1080/01932691.2020.1725543 doi: 10.1080/01932691.2020.1725543[72]Skaug M J, Mabry J N, Schwartz D K. J Am Chem Soc, 2014, 136(4): 1327−1332 doi: 10.1021/ja407396v[73]Walder R, Nelson N, Schwartz D K. Phys Rev Lett, 2011, 107(15): 156102 doi: 10.1103/PhysRevLett.107.156102[74]Dong C, Ren J. Electrophoresis, 2014, 35(16): 2267−2278 doi: 10.1002/elps.201300648[75]Wang S Q, Chang H C, Zhu Y X. Macromolecules, 2010, 43(18): 7402−7405 doi: 10.1021/ma101571s[76]Schwille P, Meyer-Almes F J, Rigler R. Biophys J, 1997, 72(4): 1878−1886 doi: 10.1016/S0006-3495(97)78833-7[77]Schaeffel D, Staff R H, Butt H J, Landfester K, Crespy D, Koynov K. Nano Lett, 2012, 12(11): 6012−6017 doi: 10.1021/nl303581q[78]Goossens K, Prior M, Pacheco V, Willbold D, Mullen K, Enderlein J, Hofkens J, Gregor I. ACS Nano, 2015, 9(7): 7360−7373 doi: 10.1021/acsnano.5b02371[79]Muller C B, Loman A, Pacheco V, Koberling F, Willbold D, Richtering W, Enderlein J. Epl, 2008, 83(4): 46001[80]Price E S, Aleksiejew M, Johnson C K. J Phys Chem B, 2011, 115(29): 9320−9326 doi: 10.1021/jp203743m[81]Torres T, Levitus M. J Phys Chem B, 2007, 111(25): 7392−7400 doi: 10.1021/jp070659s[82]Masuda A, Ushida K, Okamoto T. J Photoch Photobio A, 2006, 183(3): 304−308 doi: 10.1016/j.jphotochem.2006.06.040[83]Chen K, Zheng K K, Xu G F, Yang J F, Zhao J. Macromolecules, 2019, 52(10): 3925−3934 doi: 10.1021/acs.macromol.9b00025






















 我要推广仪器
我要推广仪器
 下载APP
下载APP