比如!C19出峰时间28.608、C20出峰时间为30.23,目标化合物出峰时间35.374,计算目标化和物的保留指数
样品和目标物的保留时间,目标离子都对的上,是不是还要看丰度比才可以判定?那丰度比的范围和标准上的相差多大才可以判定是否含有目标化合物呢?譬如名称保留时间/min选择离子m/Z丰度比DBP5.23149,150,223,2051000:92 : 47 : 43谢谢指教!!!
我想问下如果拿到一个新的目标化合物之后,怎么样才能选择一根合适的柱子来分析了?非常感谢!
目标化合物在旧柱子中拖尾,换用同类型新柱子中有所改善但是仍然呈现明显的拖尾。不知道是什么原因?有可能是因为样品稀释用甲醇-水,而流动相使用的是乙腈-水吗?样品稀释有必要换成乙腈-水吗?
我单位的GCMS是热电的,大约有一年没有用了,最近重新开始用。但是做标样时测不出目标化合物,方法是以前的方法。柱子是新的。工程师说离子源也没有问题。现在不知道是什么原因,请大家帮我分析分析。谢谢了
同一份储备液,工作溶液每天做实验现配先用。查了一些文献也没有报道这个目标化合物快速降解的问题,倒是有一篇说可能会转化,但是转化了的代谢物也有相同的母离子。应该如何去验证是什么问题?尴尬的是,方法开发都完成了,线性当时也是确认了范围可以做到,结果方法验证做到一半发现目标化合物在LQC下出不来了。如果需要调整线性的话,那就很麻烦了
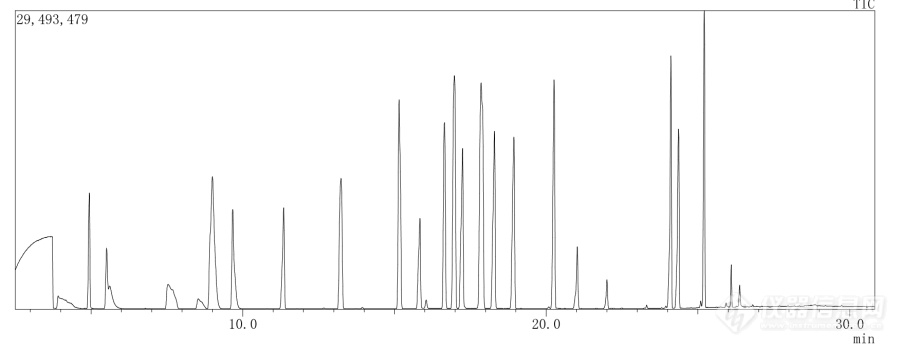
HJ 734-2014方法测固定源的挥发性有机物的方法中只给出了定量离子,定性离子都没有给出来,各位大神有没有做过这个的,有能帮忙找一下各目标化合物的定性离子,我已经用尽各种资源,实在是找不到了,大家帮忙找一下吧,另外我做出来的谱图是下边这样的,大家的做出来的是什么样的呢?[img=,690,266]http://ng1.17img.cn/bbsfiles/images/2018/06/201806121528414764_5603_3027973_3.png!w690x266.jpg[/img]
问:我想问一下保留指数的计算在没有正构烷烃的情况下,是怎么去进行计算的。======================================我答:那就不容易计算了。可以从已知的其它化合物推测,可以用脂肪酸甲酯代替测定。问:我用GC-MS测定了C8-C20,但是我的目标化合物的保留时间超过了C20的出峰时间,这样的情况是真么计算的。======================================我回复:请问C20和你的目标物的保留时间是多少?你也可以发个帖子,回复更方便。问:C20时间30.23,目标化合物时间:40.11请问您认为能计算保留指数吗?怎样计算?
求助关于TVOC的问题,按照GB5032-2001的应该程序升温到250度,计算从第一个峰开始到最后一个峰结束,TVOC的结果应该加上甲苯的测定值,而GB/T18883-2002 中计算正己烷到正十六烷之间的峰,其余峰不计在内,那我们在这中间会有很多的杂峰或者是噪音,或者像柱流失什么的,怎么判断它们是不是目标化合物,我该对哪些峰进行积分,具体的积分方法是什么,希望各位专家给予答复,希望能详细点,谢谢大家了!!!
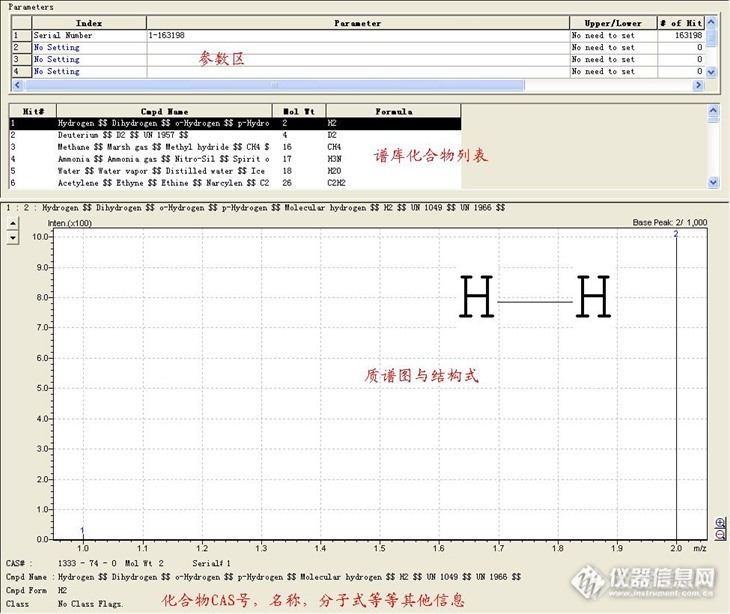
今天搜帖子,无意中发现了以前的一个求助帖,“主题:【求助】可以从gcms自带的化合物表查询目标化合物吗?如何操作?”从跟帖来看,问题还没解决。不知道楼主现在文件解决没。现在我就把方法写出来,希望楼主能看见。也希望能帮到有需要的版友。1。首先打开工作站,窗口菜单,选择show window下面的library editor,进入谱库编辑窗口http://ng1.17img.cn/bbsfiles/images/2013/06/201306261419_447816_1060664_3.jpg2。file菜单,选择open libraryhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306261419_447817_1060664_3.jpg3。在弹出的窗口中,选择一个谱库,可以是商业谱库,也可以是自建的。http://ng1.17img.cn/bbsfiles/images/2013/06/201306261420_447818_1060664_3.jpg4。以NIST05谱库为例,打开谱库后,可以看见有四大区域,具体见下图http://ng1.17img.cn/bbsfiles/images/2013/06/201306261432_447828_1060664_3.jpg5。想要查询某一种化合物的质谱图,点击参数区中的serial number,在下拉菜单中选择合适的检索参数http://ng1.17img.cn/bbsfiles/images/2013/06/201306261420_447819_1060664_3.jpg有不同的参数可供选择,常用的有分子量,分子式,名称,CAS号等。6。以最常用的CAS号为例。例如要查询3-Heptanol,CAS:589-82-2,选择“CAS Number”,在后面输入CAS号,点击检索http://ng1.17img.cn/bbsfiles/images/2013/06/201306261420_447820_1060664_3.jpg7。点击检索按钮后,检索结果出现。如果没有此化合物的质谱图,也会弹出提示,“No Hit Compound”http://ng1.17img.cn/bbsfiles/images/2013/06/201306261421_447821_1060664_3.jpg
物质的出峰大小也即峰面积或峰高,最明显的是和物质的浓度大小有关系,浓度越高响应越大,峰面积越大,现在想问的是除了和浓度有关系外,还和什么因素有关系,柱子的规格?检测器?色谱条件?同样的仪器,同样的色谱条件,不同品牌相同型号的柱子,分析同样浓度的目标化合物,响应值即峰面积相差很大,是什么原因呢?
在这个视频中,各位观众可以看到怎么利用安捷伦chemstation c版工作站的智能报告功能显示内标法目标化合物与内标物的面积比值
[font=宋体]链接:[/font]https://bbs.instrument.com.cn/topic/7815813[font=宋体]问题描述:[/font][font=宋体]固相萃取吸附剂与目标化合物之间的作用机理是什么?[/font][font=宋体]解答[/font][font=宋体]:[/font][font=宋体]固相萃取主要是通过固相吸附剂上的官能团与目标化合物的官能团之间的作用力来保留[/font]/[font=宋体]吸附的,这种作用力可分为非极性作用力、极性作用力、离子作用力、共价作用力等。[/font]a)[font=宋体]非极性作用力:对于键合硅胶及聚合物吸附剂而言,非极性作用力产生于固相萃取材料官能团上的碳氢键与目标化合物的碳氢键之间,常常被用于从样品基质中吸附分离具有非极性结构的目标化合物。如[/font]Welchrom-C18[font=宋体]、[/font]Welchrom-C8[font=宋体]、[/font]Welchrom-Phenyl[font=宋体]等。[/font]b)[font=宋体]极性作用力:极性作用力发生在许多固相萃取材料极性表面与样品中目标化合物的极性官能团之间,常见的具有极性作用力的吸附剂在色谱中一般都称为正相色谱吸附剂。常见的极性固相萃取材料包括:硅胶、氧化铝、弗罗里硅土及含有氰基、氨基、二醇基的键合硅胶。[/font]c)[font=宋体]离子作用力:离子作用力发生在带相反电荷的目标化合物与固相萃取吸附剂官能团之间。如强阳离子交换剂[/font]Welchrom-SCX[font=宋体]、强阴离子交换剂[/font]Welchrom- SAX[font=宋体]、弱阳离子交换剂[/font]Welchrom-WCX[font=宋体]等。[/font]d)[font=宋体]共价作用力:共价作用力发生在共价填料与目标化合物之间,共价键不易被打断,但有的官能团形成的共价键在改变溶剂环境的条件下是可逆的,如苯硼酸基。[/font]以上内容来自仪器信息网《样品前处理实战宝典》
我的意思是,比如我要检测分析一种化合物,但是我担心GCMS的化合物数据库没有这种物质的谱图,所以我想先查询有无这种物质。请问可以查询吗?如何操作?我用的是岛津gcms-qp2010
如题,各位大神,小白来请教。我的理解是需要让样品里所有的化合物都流出色谱柱,不然即使目标物全部流出色谱柱,有些干扰物就在色谱柱里,这对下一针的样品不会产生干扰吗?但是由此引出的问题是,我怎么知道干扰物什么时候出峰呢?或者检测器对它根本不响应,无法知道到底有没有干扰物就在色谱柱里。
目标化合物离子响应淹没在TIC中,如何降低背景TIC?
如题,小白来请教。我的理解是应该让所有的化合物都流出色谱柱,不然即使目标物出来,干扰物没出来,就在色谱柱里面,这样会影响下一针样品的测试。可是我又如何知道干扰物什么时候出峰呢,或者检测器对干扰物没有响应,我无法判断它是不是还停留在色谱柱里。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=182483]PAES.PDF[/url]谱图在附件,我们做PAES,我们现在用的FOSS 2055,用的是国产的玻璃纤维滤纸筒,就出现了硕大的峰,以前用FOSS的纤维素滤纸筒时没有出现过,可能是滤纸筒的问题 定量离子 定性离子DINP, 293, 150,126DNOP 279 150,261DIDP 307 167,150,141未知峰 279, 261,167,150,104样品中DINP,DIDP的峰很小,但是未知峰很大,我们监测7P,不想未知峰出,但是它的离子和目标化合物有相同的,如何屏蔽它?在报告的TIC中不出现,
您好,我现在正在使用Thermo的气质联用仪做实验,仪器自带了一个AMDIS软件,之前查文献了解到这个软件可以对谱图进行降噪和去卷积后再与谱库中的化合物比对,功能十分强大。但是我在Analyze-settings中的Libr选项卡中选择了Target Compouds Library后,它只能匹配出很少的目标化合物,而用NIST数据库搜索却能匹配出很多化合物。这样使得我在Generate report的时候也只有匹配出的少量化合物才有峰面积信息,所以老师我想问下您,软件可以实现对谱图中的所有保留时间下的峰面积进行积分,然后将这些积分信息导出吗?

[font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#333333]在做已知化合物校正时,设置好数据处理方法参数之后,点击重新处理,软件却出现了这样的提示:[/color][/size][/font][font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#333333][img=,690,88]https://ng1.17img.cn/bbsfiles/images/2021/08/202108081314071986_2628_3005249_3.jpg!w690x88.jpg[/img][/color][/size][/font][font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#333333]找不到我的目标化合物?那还怎么得到计算含量结果?[/color][/size][/font][color=#333333][font=-apple-system, BlinkMacSystemFont, &][size=15px]其实这个问题,跟我们设置的处理方法项下[b]化合物识别参数[/b]有关。[/size][/font][/color][color=#333333][font=-apple-system, BlinkMacSystemFont, &][size=15px]我们在做化合物识别的时候,需要填写化合物的[b]名称[/b]和[b]保留时间窗口[/b]的参数:[/size][/font][/color][font=-apple-system, BlinkMacSystemFont, &][color=#333333][font=-apple-system, BlinkMacSystemFont, &][img=,690,136]https://ng1.17img.cn/bbsfiles/images/2021/08/202108081314599091_947_3005249_3.png!w690x136.jpg[/img][/font][/color][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333][size=15px]这里面,[b]绝对RT窗口和相对RT窗口[/b]就是用来确认目标峰出峰范围的,只要样品中的色谱峰RT在时间窗口范围内,软件就可以正确识别出目标峰。[/size][/color][/font][font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#333333][b]那么这个范围是如何规定的呢?[/b][/color][/size][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333][size=15px][b]识别窗口是由预期保留时间的相对和绝对窗口来决定的,比如说,预期RT是1 min,绝对RT窗口设为[b]0.2 min[/b],相对RT窗口设为10%(注意,软件截图上可以看到,相对窗口的单位是%),我们计算得到,1min×10%=0.1 min,即相对RT窗口为[b]0.1 min[/b]。那么,窗口范围就是,预期RT左右两侧各0.2+0.1=[b]0.3 min[/b]的范围。[/b][/size][/color][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333][size=15px][b][/b][/size][/color][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333][size=15px][b][img=,690,471]https://ng1.17img.cn/bbsfiles/images/2021/08/202108081315509814_8081_3005249_3.png!w690x471.jpg[/img][/b][/size][/color][/font][font=-apple-system, BlinkMacSystemFont, &][color=#333333][font=-apple-system, BlinkMacSystemFont, &][/font][/color][/font][font=-apple-system, BlinkMacSystemFont, &][size=15px][color=#333333][/color][/size][/font]
相同浓度的目标化合物,只是一个使用甲苯作溶剂,另一个使用异辛烷作溶剂,结果峰面积相差很大,怎么回事?(用甲苯作溶剂的目标化合物峰面积为1900000左右,而使用异辛烷作溶剂的目标化合物峰面积为2400000左右。)
[color=#444444]目标化合物峰上有好多毛刺而导致峰型不好,全扫模式,SIM模式和SRM模式皆是这样,其他峰并没有出现此种情况,检测的是多环芳烃中的萘,请问是什么原因呢?[/color]
求助,各位大神,请问一下GC-MS/MS定量分析报告中的响应比为什么不等于目标化合物的响应与内标化合物的响应的比值?还有就是报告中出现的响应因子是怎么得来的?想了很久都无法理解
按照 土壤和沉积物 挥发性有机物的测定 顶空-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 HJ741-2015 做土壤中挥发性有机物,用标准方法中标准系列的中浓度点用顶空进样,定性找谱线,响应极低,很多目标化合物甚至不出峰(做的过程完全按照标准方法,已试过改变分流比,无效),直接进同样浓度的标液没有问题,是不是可以确定是顶空部分的问题,如果是顶空部分的问题,可以从哪些方面找原因?因为是新手用旧仪器,很多基础的都不懂,请大家不吝赐教。[img=,690,260]https://ng1.17img.cn/bbsfiles/images/2019/12/201912071640136439_6227_1858968_3.png!w690x260.jpg[/img] 顶空型号HP7694,GC:安捷伦6890N,谱图见附件
在这个视频中,各位观众可以看到怎么利用安捷伦chemstation c版工作站实现利用智能报告计算内标法中多个目标化合物与内标物的面积之比
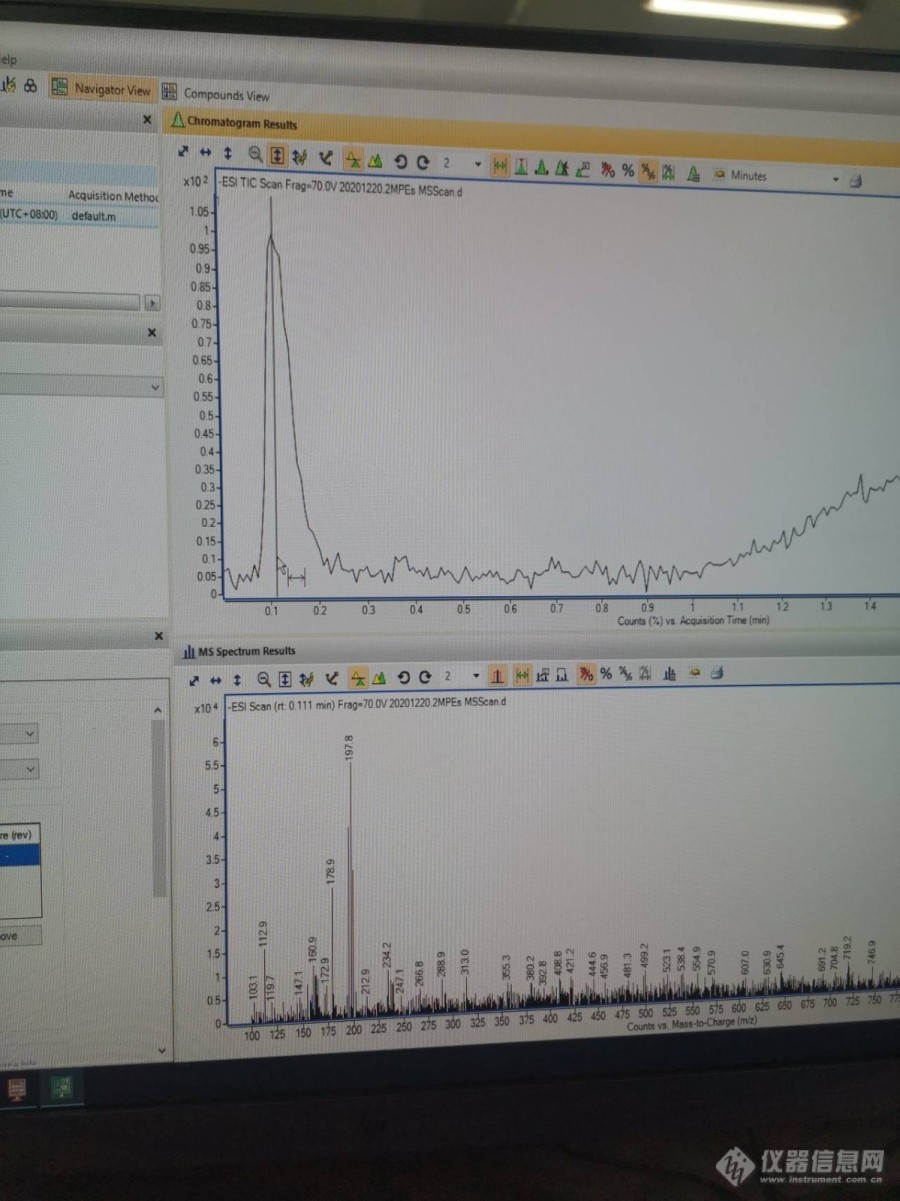
各位,大家好,刚接触[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],使用MS-SCAN扫描,得到总离子流图,如何下图,只有一个峰,目标化合物没有峰。我化合物的母离子文献上查的m/z为179.035。想请教一下大家:1.目标化和物没有峰是不是浓度太低?2. TIC图,怎么不是别人那种基部很平滑那种[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2020/12/202012192144360345_9628_5127102_3.jpg!w690x920.jpg[/img]
[color=#444444]做了2个平行样,结果发现其中一个样品中的目标化合物的离子对丰度比发生变化,出峰时间跟标准溶液是一致的。而且与另一平行样比较发现,这一样品中的定量离子响应值是一致的,但是定性离子的响应值低了10倍左右。那为什么会出现这个现象呢?前处理以及仪器条件肯定都是一样的[/color][color=#444444]另外,欧盟 2002/657文件对分析方法中的定量及确证提出的要求和规定,[/color][color=#444444]相对离子丰度 >50% >20%至50% >10%至20% ≤10%[/color][color=#444444]允许相对偏差 ±20% ±25% ±30% ±50%[/color][color=#444444]为什么要做出这样的规定呢?如果超出这个偏差范围说明什么问题,如何应对呢?[/color]
如题,在LCMS的实际测试中,发现一些硝基和氰基化合物的质谱信号不好,经常找不到目标峰。是不是这类化合物在ESI的条件下不易被离子化啊?原因又是什么呢?
群友提问: 咨询一下:用气相质谱仪定性时候,每种目标化合物定性定量离子一共选择几个合适?有人回复: 1. 有机氯,有机磷,菊酯类的2. 我们做的有机氯有机磷,菊酯类都没检出来过大家有谁知道呢?
用外标法定量后,对样品进行定量计算,可是目标化合物的保留时间和响应全都为零,但图谱是有的,这是为什么?有什么解决的办法呢?求大神指点。。http://ng1.17img.cn/bbsfiles/images/2017/02/201702211526_01_3187540_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP