同一瓶样品,重复称样、定容、手动进样8次。C18柱流动相:按药典缓冲盐:乙腈(80:20),主峰前有3个杂质峰,主峰后有一个,只是第一个杂质峰面积不稳定,上下波动。第一个杂质峰第一针峰面积156;第一个杂质峰第一针峰面积52第一个杂质峰第一针峰面积49第一个杂质峰第一针峰面积40第一个杂质峰第一针峰面积57第一个杂质峰第一针峰面积32第一个杂质峰第一针峰面积50第一个杂质峰第一针峰面积49又对另一个批次样品进样2针(重复称样)第一个杂质峰第一针峰面积 159第一个杂质峰第一针峰面积 83老师们分析下,到底是系统问题还是样品中相关杂质不稳定?又该如何调整?跪求答复!进样前基线和空白对照都很好!不同样品间都走空白,空白此杂质的积分面积只有1个单位!可以忽略1
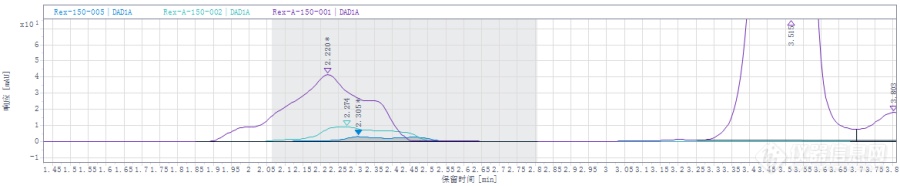
最近检测一个化合物,在2min左右有一个杂质,它的峰面积随着流动相乙腈比例减小而减小,下面是谱图,乙腈比例是40、38、36%,而且空白处在该时间段没有干扰峰,我想问下这种情况杂质峰面积该怎么确定呢?[img=,690,142]http://ng1.17img.cn/bbsfiles/images/2017/10/201710241104_01_3142880_3.png!w690x142.jpg[/img]
实验室研发的新产品即将生产,其中有一个[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]监控的步骤,需要用简单的方式来定量控制该物质的含量。杂质是异丙醚,此步是中间控制,需要将该杂质含量控制在1%以内,领导要求开发一个简便的方式来定量从而控制其含量。我想了一下,能否将产品与杂质1:1配制,溶解至100ml,那么此时杂质与产品的含量比即为杂质与产品的峰面积比。问过研发人员,中间产物中,产品大致的量他们是知道的,也就是产品的量已知。那么能否通过直接进样中间产物,观察产品与杂质的比值来判断杂质的大致含量呢?这个方式我在液相的杂质含量中其实有大致了解,但是无奈理解能力有限,其实没有太懂[b][color=#000000]加校正因子主成分自身对照法[/color][/b]与[color=#000000][b]不加校正因子的主成分自身对照法[/b]的[/color]计算方式。[b][color=#ff0000]举个例子:[/color][/b]第一种[font=&][color=#000000][size=14px][b]加校正因子主成分自身对照法[/b][/size][/color][/font]:假设我现在有杂质对照品,和产品对照品,我将两种配制成浓度已知的混合液,进样,已知校正因子(f)=(S杂质*C杂质)/(S产品*C产品)计算出校正因子,假设(f)=杂质:产品=1.2那么我现在有个中控液进样了,杂质:产品峰面积为1:10,产品在混合液中的含量差不多为50%,那么杂质含量占多少呢?杂质含量此时是6%吗?第二种[b]不[/b][font=&][size=14px][color=#000000][b]加校正因子主成分自身对照法[/b][/color][/size][/font]:这种我是完全没懂了,,,懵圈了。如果我第一种方法的理解没错的话,那么此次[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]中控方式能否用这种方式来计算杂质的大致含量呢。求教。
同一个样品正常的时候杂质峰面积小,非正常的时候杂质峰面积变大6-7倍
今天做4AA,除了产品峰外,前面还出现一个大的杂质峰,稀释两倍后重新进样,杂质峰不但没变小,反而变大,产品峰变成原来的1/2,有没有遇到这种情况的.跑过溶剂空白,没有大的峰!!请各位大虾指教~!谢了~
FID检测器里面有杂l杂质,怎么处理
做纯物质中的杂质含量,同样的标样浓度,现在杂质的响应面积好小好小,该怎么查原因?
[B][center]药品研发如何确定杂质限度[/center][/B][B]国家食品药品监督管理局药品审评中心 黄晓龙[/B] 在药品研发中,如何证实药品安全有效应该是研发人员始终关注的问题;而药品质量的稳定可控又是保证其安全有效的前提与基础。如果一个药品的质量不能达到稳定与可控,在使用时这一药品就不可能始终安全、有效,也就不能被批准上市。保证药品质量稳定可控,药品的纯度是一个重点。如何确定杂质的限度是药学研究人员与审评人员不能回避的关键问题,该限度的制订是否科学、合理,直接关系到药品的安全性与质量。药品在临床使用中产生的不良反应除与该药品本身的药理活性有关外,也有一部分与药品中所混入的其它杂质有关。例如,通过我国药学科技工作者数十年的努力,基本上确定青霉素等抗生素中的多聚物等高分子杂质是引起过敏的主要原因。所以在研发过程中一定要对药品中的杂质进行全面研究,并将杂质完全准确地控制在一个合理的范围之内。 尽管杂质限度的确定对于药品研发非常重要,但国内药品研发的现实情况并不令人乐观。从近几年的新药申报情况分析,在杂质的研究与限度确定方面存在着较多的问题,主要表现为:部分药品研究单位对杂质研究的重要性了解不深;标准中对杂质的控制不够全面与准确;制订杂质限度时考虑问题不够全面,很少考虑杂质对药品安全性的不良影响;即使在杂质的含量明显超出正常工艺所允许的范围时,也不注意对现有的处方与工艺进行必要的优化,以降低杂质的限度。◆杂质的分类 药品中的杂质一般分为三类:有机杂质、无机杂质及残留溶剂。 有机杂质是指在药品的生产与储存过程中产生的杂质,这些杂质可以是已知的、未知的、挥发性的或不挥发性的杂质,主要包括:降解产物、聚合物、原料药与辅料或内包材的反应产物、以及原料药制备过程中引入的起始原料、副产物、中间体、反应试剂、配位体与催化剂。由于这些杂质的化学结构与产品分子类似或具渊源关系,所以通常称之为有关物质。 无机杂质是指在药品的生产过程中产生的杂质,这些杂质通常是已知的,主要包括:反应试剂、配位体与催化剂、重金属或其它残留的金属、无机盐、过滤助剂、活性炭等其它物质。 残留溶剂是指在原料药及制剂的生产过程中使用的有机溶剂。 对于生产过程中引入的外来污染物,可通过“良好的生产规范”(GMP)来控制,故不属于本文所说的杂质范畴。原料药的不同晶型也不属于本文的讨论范畴。本文只谈有机杂质与无机杂质的限度确定。

http://ng1.17img.cn/bbsfiles/images/2017/10/2015102217103728_01_0_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015102217092431_01_2147443_3.jpg如题,我们产品里面有个杂质组分,平时约在0.2-0.4%(按面积百分法计算的结果),但是最近两天这个杂质峰面积变化异常,第一针达到1%,反复进样后逐渐变小,最后变为正常值(0.2-0.4%)。其他组分峰的重现性较好。空白与STD中都无此时间的出峰。我进样口隔垫、存管、进样针、洗针液都换了,相比平常为什么还这样不稳定。
做实验时,发现溶液中突然出现少量杂质,怀疑是通风橱打开引风机后引入的,有没有什么好方法可以避免这种现象,已经盖表面皿了。
如题,主成分和其他杂质未出峰。但是更换流动相体系后主成分和一部分杂质出峰了。主成分和其他杂质在该波长下均有吸收。我有必要为了其他物质出峰而改变流动相吗?如果不改变,其他未出峰的物质会对我的检测有什么影响?
做纯物质中的杂质含量,同样的标样浓度,现在杂质的响应面积好小好小,该怎么查原因?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181311]多晶表面金属杂质测定方法.doc[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181312]ASTM(F1724)多晶表面金属杂质和磷硼含量ASTM标准测定.pdf[/url]
根据IFRA的信息通讯(IL 948),考虑科技、分析方法和监管的发展,IFRA认为对其关于日用香料中无法避免的痕量杂质问题的立场,需要进行重新定位。不同的法规体系,对“痕量水平”的定义不尽相同,而且分析方法的灵敏度不断提高,物质的检出限越来越低;但是,监管框架并未发生变化,生产者应确保痕量杂质的存在不影响原料和产品的使用安全。 考虑到安全,IFRA已经对一些日用原料中含量极低的杂质成分进行了限量,这是针对某些物质的具体情况而制定的,而不是关于痕量污染物的通用规定。据信,一些国家和地区的监管也采取这种方式。 IFRA认为对众多日用香料中的所有痕量物质制定一个单一的通用限量的观点并不恰当,也无必要。只有IFRA认为确有必要对某些物质(例如,苯、甲苯、黄樟素等)进行限制时,才会制定相应的IFRA标准。 任何对痕量物质的管理研究都应建立在各成员企业依据法规要求对单个物质的安全考量和预期使用的基础上。
一直烦恼配杂质标液的问题,因为玻璃仪器中的Na 和Si很容易污染标液,但是用塑料容量瓶的话,发现移液管靠在塑料壁上往下流后,残留的液体比靠在玻璃管壁上的要多,虽然只是一点,可是浓度还是会有一点点的影响吧。不过我现在都要求用塑料瓶子去配杂质标液,受污染的程度会减少一点。大家是怎么避免杂质标液被污染的?
现有一个产品检测项目,按照药典要求,各杂质计算需要采用杂质溶液自我稀释的方法来计算,由于按照药典全套检测,需要大量人力物力。 想请教一下:从GMP角度讲,是否可以直接采集杂质溶液,通过读取色谱图上各杂质的结果来判定盖批次样品是否可以用于小批次混合
请教大家,锆合金中的杂质该怎么做,如何避免锆基体干扰,能否将锆和杂质元素分离?
国标GB/T 9996.2-2008 《棉及化纤纯纺、混纺纱线外观质量黑板检验方法 第2部分:分别评定法》中,要求在数棉结杂质时,将“浅蓝色底板”插在试样和黑板之间。那么,这个浅蓝色底板是标准物质吗?
我们用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]面积归一化法测定胺类物质含量,现发现杂质含量偏低(原来正常后突然变低,在其它仪器上测定结果正常),载气压力变大时杂质含量有所增大,不知是何原因,请高手指教!谢谢!
采用面积归一化法测定高分子杂质,但系统适用性的对照品溶液峰面积rsd达到9%以上,这样会影响杂质定量么,有人说峰面积归一化法不用对照品峰面积,不用管,有做过的么?(对照品溶液和杂质不是一个物质)
在有关物质方法开发过程中,会考察一些杂质,与主成分混成一个系统适用性溶液,在同一波长下检测,但有些杂质可能与主成分响应不一致,就需要引入校正因子,但是本人没有经过这方面的培训和学习,对这方面的规范操作不是很明白,希望有这方面经验的老师和同仁们,不吝赐教。问题:1、对于考察的杂质而言,方法验证时,是所有杂质都需要做校正因子,还是有选择的?比如只选择与主成分响应不一致的杂质进样校正因子测定?2、如何判断杂质与主成分响应值不一致?是在DAD检测器下的图谱中,看峰的紫外扫描图吗?3、杂质校正因子与相对校正因子的计算公式?我们目前是将已知杂质的两个浓度(一个是约0.1mg/ml浓度的,另一个是0.01mg/ml)各进3针,取平均值,校正因子=峰面积/浓度;相对校正因子=杂质校正因子/主成分的校正因子。
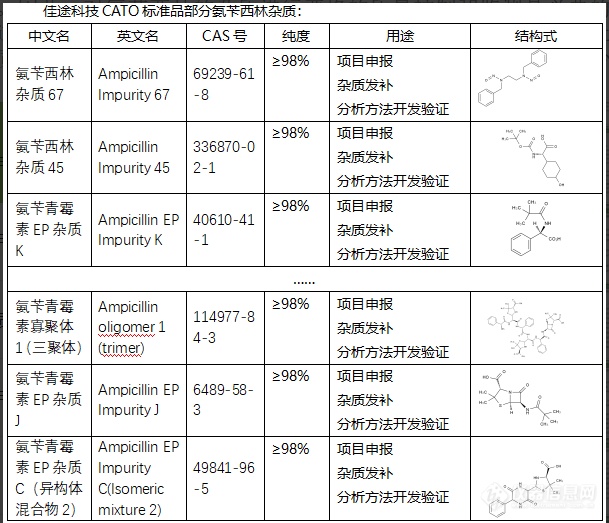
氨苄西林的杂质对于其质量、效力和安全性非常重要。一些杂质可能对药物的安全性和疗效产生负面影响。杂质的存在可能引发药物不良反应,如过敏反应,或者降低药物的效力。因此,对氨苄西林的杂质进行严格的质量控制和监测是必要的。质量控制不仅包括在生产过程中对杂质的检测和去除,还包括对贮存条件的监控,以防止在贮存过程中产生新的杂质或使现有的杂质浓度升高。CATO标准品对氨苄西林杂质的研究也有利于优化制药工艺,从而提高药物的质量,并减少不良反应和副作用的危险。这对于保证药物的治疗效果和患者安全性至关重要。[img=,609,523]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052057087895_2740_6381668_3.png!w609x523.jpg[/img]
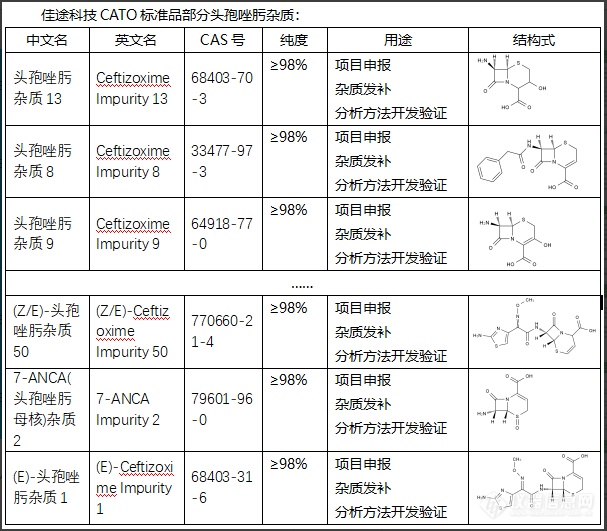
有关头孢唑肟杂质的作用,以下是要注意的一些可能性:1.负面作用:过多的杂质可能导致药物效力下降,并可能引发不良反应或副作用。例如,有些杂质可能导致过敏反应。2.毒性:某些杂质可能具有毒性。例如,某些杂质可能具有致癌性。3.影响药效:杂质可能会影响药物的生物利用度,即药物进入体内后能达到预期药效的能力。CATO标准品药品生产中的质量控制步骤非常重要,目的就是要尽可能减少杂质的存在。任何药品都必须经过严格的质量检测,确保其安全有效。[img=,607,531]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041449269442_4660_6381668_3.png!w607x531.jpg[/img]
想请教一下各位老师GC色谱上的某峰我怀疑是杂质A,它保留时间和A的是一样的,往里面加A这个杂质峰面积确实变大了(回收率是98%),能不能证明这个峰就是A的峰?还是说有可能是其他极性相似的杂质?类推一下,想问问加样实验之后增加了的那个峰一定是你加进去的那个杂质的吗?(在确定已经含有该成分之后)A是个酸,之前走其他柱子可以检出主成分的其他杂质,换了极性柱之后只剩下主峰和疑似A的峰检测器是FID
药典描述:分别精密量取供试品溶液、对照品溶液和系统适用性溶液各1μl注入气相色谱仪,记录色谱图,按内标法以峰面积计算,供试品含二甘醇与乙二醇均不得过0.025%;如有其他杂质,扣除内标峰按归一化法计算,单个未知杂质不得过0.1%;杂质总量(包含二甘醇、乙二醇)不得过1.0%。其中杂质总量应该是按面积归一法计算,还是由其他杂质+二甘醇、乙二醇(内标结果)?请问生产厂家和使用单位都是如何计算的?杂质总量前为分号!!!
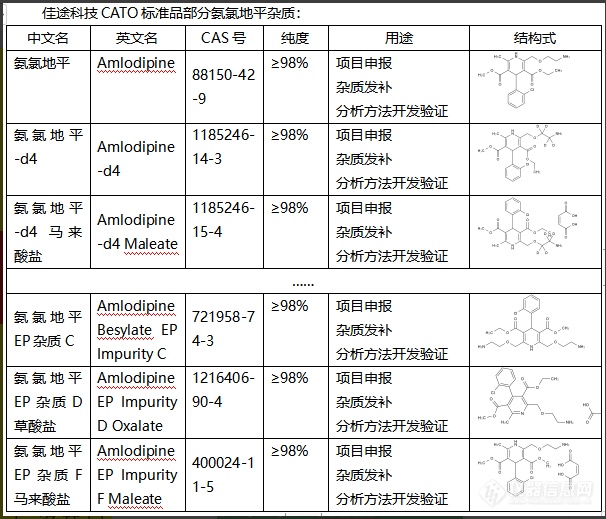
氨氯地平是一种常用的抗高血压药物,对于其中的杂质,氨氯地平杂质其作用主要体现在以下几个方面:1. 质量影响:杂质会影响氨氯地平的纯度和稳定性,可能会导致药品质量下降。2. 安全性影响:杂质可能会产生一些未知的副作用和毒性反应,影响药品的安全性。3. 药效影响:杂质可能会干扰氨氯地平的药效,使得药物的治疗效果降低。4. 法规因素:食品药品监管部门对药品中的杂质有严格的限制标准,过多的杂质可能会导致药品不能上市。CATO标准品对于氨氯地平这类药物的生产,控制和降低杂质的含量是非常注重的。[img=,606,519]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041354066781_2922_6381668_3.png!w606x519.jpg[/img]
ICP测金属镍里面的杂质,要怎么溶样?
大家好!不知道大家有没检测溶剂里面的杂质?最近在测试碳酸二甲酯中的杂质,用其他溶剂稀释,却引入其他杂峰,搞得杂质都没测试出来,扣过背景后,都查不出有什么杂质峰。大家有什么好的方法来检测溶剂里面的杂质?http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
因为换了工作,对现在公司的一些处理方式很不理解,问了一些同事,发现他们也不懂,只知道一直以来是按照那样用。可是我认为那样不对,遂发上来和大家讨论。以下说的都是我现在所在公司的做法:1.在进样之前,先进一针LOQ(定量限)溶液,如果LOQ中特定峰S/N(信躁比)=10或者连续进三针,峰面积RSD=10%(根据具体而定),则认为LOQ通过,否则不通过,不能进行后面的分析。2.在LOQ通过的情况下,进样品,然后用面积归一化法,对图谱进行分析,据此得出各杂质的含量,前面进的LOQ得到的数据不参与结果的计算。3.对于色谱图中,小峰的处理是这样的,峰面积百分比=LOQ(比如0.1%,将样品溶液稀释1000倍)则积上,否则不积。对于这样的做法,我主要有以下几个疑问:1.LOQ不是在做方法验证的时候才需要做吗?而且药典(包括USP)上明确写了,除了杂质定量外,都可以不做。那么每次都做个LOQ,有意义,有必要吗?2.将样品溶液稀释1000倍,得到的是浓度,怎么直接把它给转换成了峰面积比?这样科学吗?当然了,不考虑响应值的不同,即不引入校正因子。3.想问一下,大家对小峰是怎么处理的呢?一个小峰到底积不积上,根据什么来判断呢?我们以前基本是根据经验的。4.药典上不是说用面积归一化法对杂质进行定量分析是很不准确的吗?那仍然这样分析是否合理。5.药典上说的杂质定量都是与自身比对(与另一低浓度),那么我们这样的做法科学吗?希望大家不吝赐教!
[b] [size=18px] 现有一个品种,主峰后有一个未完全分离的杂质,定量方法是主成分自身对照法,结果发现不同的积分参数对杂质峰面积影响很大,不同积分方式直接导致合格或超限! 请大神来判断一下,最合理的最能反映真值的积分方法是哪种? 具体见下图:[/size][img=966a4957e5ea5d0956f4d63083fbe61f.jpg]https://pic.ouryao.com/data/attachment/forum/202211/17/094745w5xp5epro14ipe3z.jpg.thumb.jpg[/img][font=-apple-system, BlinkMacSystemFont, &][size=16px][color=rgba(0, 0, 0, 0.85)]目前我用的是垂线积分,结果杂质按自身对照法算超过限度,而换成谷谷积分,则杂质含量没有超限,但我个人认为谷谷积分不够准确,属于人为减少了杂质峰面积,个人觉得第4中积分方式,把主峰撇去,指数拟合出完整的杂质峰型才是最准确的,但不知道变色龙软件有没有这种积分功能?其次是垂线积分,是比较接近真实杂质含量的积分。谷谷积分直接把基线上方一大啪峰面积给去掉了,对于自身对照法来说,结果肯定是偏小的,个人觉得不准确。求大神发表意见。[/color][/size][/font][/b]







 我要推广仪器
我要推广仪器
 下载APP
下载APP