酶切反应建议一个版友提供的简述版的酶切反应建议,还是很不错的,很适合初步做酶切反应者。一、 建立一个标准的酶切反应二、 选择正确的酶三、 酶 四、 DNA 五、 缓冲液六、 反应体积七、 混合 八、 反应温度九、 反应时间 十、 终止反应 十一、贮存 十二、稳定性 十三、对照反应 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=158046]酶切反应建议[/url]
求够酶反应器 联系电话 02783922509 联系人 王先生
聚合酶链式反应的主要用途在那些领域?
聚合酶链反应 现已报道用多种不同条件通过PCR扩增DNA片段,现归纳如下。当对哺乳动物DNA 进行实验时,我们用的反应总体积为50μl,内含50-500ng基因组DNA.反应缓冲液: 67mM Tris.HCl,pH8.8,6.7mM MgCl2,16mM(NH4)2SO4,10mMβ羟基乙醇和10%二甲 基亚砚。反应液同时分别含有75pmol四种脱氧核苷三磷酸、每一种寡核苷酸引物为 50pmol.混匀样品后,加入一个单位的Taq DNA聚合酶,100μl矿物油覆盖于反应溶 液的上层以防蒸发。反应样品于93℃保温1分钟使DNA变性,然后如下所述在60℃到 70℃之间放置1-2分钟。扩增后,将水相转入另一支试管,酚抽提及乙醇沉淀。用 50μl非变性胶载样缓冲液悬浮在PCR扩增过程中出现的一些问题以及建议如下 一、错误的PCR扩增片段:使用简便的EB染色检测在变必梯度胶中的扩增DNA,经PCR 扩增应该合成单一的DNA.不幸的是反应产物偶尔比预料的要复杂得多;例如形成除靶 DNA之外的许多不同大小的DNA,有时这些非靶DNA片段占产物的绝大部分。在此情况 下,额外产笺DNA干扰了在DGGE上对靶片段的分析。我们采取以下几个步骤为防止上 述问题的出现: 1、在PCR过程中避免产生非靶DNA片段的方法之一是使反应在尽可能高的温度下进 行。反应在低温下(45-55℃)退火有可能使引扩增基因组DNA上的其它区域而不是靶片 段。很可能引物不能完全与非靶DNA序列互补,但经PCR最初合成反应之后,这些新的 产物在随后的扩增循环中成为退火良好的模板。因此,我们一般在尽可能高的温度下 退火,通常是55-65℃,然后在可能的最高温度下延伸,一般为60-72℃。对于每一对 寡核苷酸温度的选择是根据经验来决定的。2、选择比较长的寡核苷酸引物(25-30个核苷酸而不是20个,不包括GC发夹序列) 有时能改善反应的特异性,减少非目的产物。较长的引物同时可增高退火/延伸的温 度,而这也增加了反应的特异性。实际上,30个核苷酸长的引物在PCR过程中可直接 在两个温度之间(通常是70-72℃和93℃)进行循环反应,而不需要通过退火步骤,从 而有助于防止非物异的带产生。
[em0901]三聚氰胺能和哪种酶反应?意思是用哪种酶和三聚氰胺来反应,检测三聚氰胺的含量!
定义 聚合酶链式反应简称PCR(英文全称:Polymerase Chain Reaction), 具体内容点击: 聚合酶链式反应,简称PCR。聚合酶链式反应,其英文Polymease Chain Reaction(PCR)是体外酶促合成特异DNA片段的一种方法,由高温变性、低温退火及适温延伸等几步反应组成一个周期,循环进行,使目的DNA得以迅速扩增,具有特异性强、灵敏度高、操作简便、省时等特点。它不仅可用于基因分离、克隆和核酸序列分析等基础研究,还可用于疾病病的诊断或任何有DNA,RNA的地方.聚合酶链式反应(Polymerase Chain Reaction,简称PCR)又称无细胞分子克隆或特异性DNA序列体外引物定向酶促扩增技术。 由美国科学家PE(Perkin Elmer珀金-埃尔默)公司遗传部的Dr. Mullis发明,由于PCRPCR技术在理论和应用上的跨时代意义,因此Mullis获得了1993年诺贝尔化学奖。 技术原理 DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶与启动子的参与下,根据碱基互补配对原则复制成同样的两分子挎贝。在 实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,并设计引物做启动子,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制。 但是,DNA聚合酶在高温时会失活,因此,每次循环都得加入新的DNA聚合酶,不仅操作烦琐,而且价格昂贵,制约了PCR技术的应用和发展。发现耐热DNA聚合同酶--Taq酶对于PCR的应用有里程碑的意义,该酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也大大降低了成本,PCR技术得以大量应用,并逐步应用于临床。 工作原理 类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右一定时 间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。
最近,在做一个工艺实验:主要是利用鲜叶中的多酚氧化酶酶促氧化生产茶色素。中间需要在线取样分析含量变化。但是因为溶液里面残存有酶液,如果不将酶失活或者处理的话,反应还会继续,那如何终止酶反应呢?目前我们的做法是:1. 取出后,用普通滤纸过滤,放置零下20度冰箱中保存,直至开始分析。2. 取出,加入有机溶剂,如乙醇或者乙腈,将反应终止,分析!有个小问题:在1中,用滤纸过滤,感觉酶一般比较大,可以被截留?不知道是不是这样呢?大家还有什么好 的办法没有??
前一阵子一直在做双酶切质粒重组,失败了 很多次,不过很快改善了 实验方法,用2周重组了 14个质粒。现就自己的体会,结合丁香园战友的宝贵经验,谈一下质粒重组的一些个人经验。1. 回收PCR产物 在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的 选择非常重要,尽量选择粘端酶切和 那些酶切效率高的限制酶,如BamHI、HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照:http://img.dxy.cn/upload/2006/08/13/31219184.pdf。双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒。酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/微升,在除外酶降解的 因素外,该酶可分解15 μg的DNA,而一般从1-4 ml菌液提出的 DNA约为3 μg,而PCR纯化后的产物(50体系)约为3 μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。2. 酶切、回收后的PCR产物与载体的连接摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03 pmol,后者取0.3 pmol。pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1 pmol 1000 bp DNA=0.66 μg,如载体是5380 bp,则0.03 pmol为0.03×5.38×0.66=0.106524 μg。测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0,另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的 MARKER每个条带约50 ng。连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。3. 转化a. 全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。b. 42℃加热45秒钟后,再在冰中放置1分钟。c. 加入890 μl AMP阴性培养基,37℃振荡培养60分钟。取100 μl铺板。也可离心后余100 μl。几个非常重要的问题1. 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解,为保险起见,一般连接3小时,16度。2. 对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干。3. 对照的设立:为验证双酶切是否成功,可做如下对照:A 酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性,如两管均成线性可初步判断双酶切成功。做转化时也要进行对照。设4个:A. 即拿双酶切的质粒产物也进行连接反应,这个对照可进一步看双酶切是否成功,如果长出克隆,说明很有可能只进行了单酶切,如没长出克隆,则证明双酶切成功,当然要保证感受态,培养基、连接酶都'正常'的情况下。B. 酶切过的未进行连接反应的双酶切产物,进行转化,这一步可以证明是否有残留的未被任何酶切的原始质粒。C. 设原始质粒为对照,意为检测整个操作过程中是否有误。D.AMP阴性板上用同一批感受态细胞铺板20微升足够,检测感受态状况。4. 所有的试剂切记低温保存 一步一个脚印,不要偷懒,图省事最后却更费事,注意设立对照。经PCR鉴定,克隆90%-100%的阳性率,所以在后面的 挑克隆中,我只挑选4个就足够了。然后双酶切鉴定,测序。
聚合酶链式反应是一种用于放大扩增特定的DNA片段的分子生物学技术,它可看作是生物体外的特殊DNA复制,PCR的最大特点,是能将微量的DNA大幅增加。因此,无论是化石中的古生物、历史人物的残骸,还是几十年前凶杀案中凶手所遗留的毛发、皮肤或血液,只要能分离出一丁点的DNA,就能用PCR加以放大,进行比对。这也是“微量证据”的威力之所在。由1983年美国Mullis首先提出设想,1985年由其发明了聚合酶链反应,即简易DNA扩增法,意味着PCR技术的真正诞生。到如今2013年,PCR已发展到第三代技术。1973 年,台湾科学家钱嘉韵,发现了稳定的Taq DNA聚合酶,为PCR技术发展也做出了基础性贡献。PCR(聚合酶链式反应)是利用DNA在体外摄氏95°高温时变性会变成单链,低温(经常是60°C左右)时引物与单链按碱基互补配对的原则结合,再调温度至DNA聚合酶最适反应温度(72°C左右),DNA聚合酶沿着磷酸到五碳糖(5'-3')的方向合成互补链。基于聚合酶制造的PCR仪实际就是一个温控设备,能在变性温度,复性温度,延伸温度之间很好地进行控制。Khorana (1971)等最早提出核酸体外扩增的设想:“经DNA变性,与合适的引物杂交,用DNA聚合酶延伸引物,并不断重复该过程便可合成tRNA基因。”但由于当时基因序列分析方法尚未成熟,热稳定DNA聚合酶尚未报道以及引物合成的困难,这种想法似乎没有实际意义。加上70年代初分子克隆技术的出现提供了一种克隆和扩增基因的途径,所以,Khorana的设想被人们遗忘了。1985年,Kary Mullis在Cetus公司工作期间,发明了PCR。Mullis要合成DNA引物来进行测序工作,却常为没有足够多的模板DNA而烦恼。1983年4月的一个星期五晚上,他开车去乡下别墅的路上,猛然闪现出“多聚酶链式反应”的想法。1983年12月,Mullis用同位素标记法看到了10个循环后的49 bp长度的第一个PCR片段;1985年10月25日申请了PCR的专利,1987年7月28日批准(专利号4,683,202 ),Mullis是第一发明人;1985年12月20日在Science杂志上发表了第一篇PCR的学术论文,Mullis是共同作者;1986年5月,Mullis在冷泉港实验室做专题报告,全世界从此开始学习PCR的方法。DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解旋成单链,在DNA聚合酶的参与下,根据碱基互补配对原则复制成同样的两分子拷贝。在实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,加入设计引物,DNA聚合酶、dNTP就可以完成特定基因的体外复制。但是,DNA聚合酶在高温时会失活,因此,每次循环都得加入新的DNA聚合酶,不仅操作烦琐,而且价格昂贵,制约了PCR技术的应用和发展。耐热DNA聚合酶--Taq酶的发现对于PCR的应用有里程碑的意义,该酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也大大降低了成本,PCR技术得以大taq酶taq酶量应用,并逐步应用于临床。PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在72℃、DNA聚合酶(如TaqDNA聚合酶)的作用下,以dNTP为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环变性--退火--延伸三过程就可得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。
前一阵子一直在做双酶切质粒重组,失败了 很多次,不过很快改善了 实验方法,用2周重组了 14个质粒。现就自己的体会,结合战友的宝贵经验,谈一下质粒重组的一些个人经验。1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的 选择非常重要,尽量选择粘端酶切和 那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/微升,在除外酶降解的 因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的 DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。2、酶切、回收后的PCR产物与载体的连接摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03pmol,后者取0.3pmol。pmol为单位的DNA转换为为µg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为µg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为0.03×5.38×0.66=0.106524µg。测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的 MARKER每个条带约50ng。连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。3、转化:a、全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。 b、42℃加热45秒钟后,再在冰中放置1分钟。 c、加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl
酶反应,产物为3-羟基丙酸,HPLC检测,有机酸柱,流动相为5mM硫酸。之前用10%硝酸使酶变性,离心后取上清进样,但是硝酸会产生一个很大的峰和产物峰重叠,请问该体系有什么合适的前处理方法去除酶么?谢谢!
蒋华良教授的科普文章《红烧肉中的著名化学反应》,受到了广大读者的喜爱,网络上很多人学习。许多人对不用酱油烧红烧肉很感兴趣,因此,蒋教授修改了文章,增添了有关焦糖(糖饴)制作相关化学反应,也介绍了他从小学的独门糖饴制作方法。红烧肉中的著名化学反应--美拉德反应(更新版)蒋华良(中国科学院上海药物研究所)前一段时间,央视8台播出广告--我们恨化学,引起了广泛关注和热议。这件事引起了我的深刻反思:作为一名化学研究工作者,没有尽力去正面宣传化学,向老百姓科普化学。从今天起,我将抽空写些关于化学的科普小品文,让更多的人了解化学,公正地看待化学。我们每天的衣食住行离不开化学。例如,我们每天要吃油盐酱醋糖,这些常用佐料的制造,均离不开化学。有人会认为糖是从甘蔗和甜菜等植物中榨取的,以为用不上化学。其实,白糖的制造过程用到的过滤、蒸发、结晶等技术均是常用的化学技术,食盐的制造同样要用到这些技术,酱油和醋的生产主要靠发酵,其中发生了很多生物化学反应,也要用到过滤和精制等传统化学技术。有一门化学分支,叫食品化学,与我们的饮食关系极大。食品化学是系统研究食品的化学组成、结构、性质以及食品加工和贮藏过程中发生的化学变化的科学。食品在加工工程中会发生很多化学反应,有些化学反应非常有趣。红烧肉是我国老百姓喜爱的家常菜,几乎没有人不喜欢吃红烧肉的。做红烧肉时通常要加白糖和料酒(黄酒),一般认为氨基酸与乙醇发生酯化反应,生成氨基酸乙酯,这一反应显示了料酒去腥的作用,红烧肉的香味主要也是氨基酸乙酯的功劳。其实,红烧肉的香味主要是白糖的功劳。今天介绍做红烧肉的过程中发生的一个化学反应,这一化学反应是红烧肉色泽、香味和好味道的主要因素。1912年法国化学家L.C.Maillard发现氨基酸或蛋白质与葡萄糖混合加热时形成褐色的物质。后来人们发现氨基酸或蛋白质能与很多糖反应,这类反应不仅影响食品的颜色,而且对食品的香味也有重要作用,人们将此反应称为美拉徳(Maillard)反应或非酶褐变(nonenzymaticbrowning)反应。只要温度不高,如做红烧肉,这种反应产生的褐色物质无毒,且香气扑鼻,色泽诱人,是红烧肉、红烧鱼等成为美食的功臣。不同的氨基酸与不同的糖反应,能产生不同的香味。例如,亮氨酸与葡萄糖在高温下反应,能够产生令人愉悦的面包香。红烧肉的香味比较复杂,还不知什么氨基酸与糖反应的,可能是多种氨基酸与多种糖反应的产物。美拉德反应还促进了香料化学的发展,该反应在香精领域中的应用打破了传统的香精调配和生产工艺的范畴,产生了一种全新的香料生产技术,尤其在调味品行业应用广泛。该反应所形成的香精能产生天然肉类香味的逼真效果。美拉德反应的机制还不十分清楚,1953年Hodge对美拉德反应的机理提出了系统的解释,可分为三个阶段:初期、中期和末期。初期是氨基酸的氨基与糖的羰基发生亲核加成反应生成席夫碱,席夫碱环化形成氮代糖基胺,经阿姆德瑞分子重排反应,生成烯醇式和酮式糖胺。中期是烯醇式和酮式糖胺在酸性条件下经1,2-烯醇化反应,生成羰基呋喃醛,在碱性条件下经2,3-烯醇化反应,产生还原酮类和脱氢还原酮类化合物。这些多羰基不饱和化合物通过斯特勒克(Strecker)降解反应,生产醛类、吡嗪类化合物和一些容易挥发的化合物,这些化合物能产生特殊的香味。最后阶段的机制非常复杂,多羰基不饱和化合物进行缩合、聚合反应,产生褐黑色的类黑精物质。类黑精物质是红烧肉色泽的物质基础,控制糖的量和温度,缩合、聚合反应的程度不同,产生不同的类黑精物质,红烧肉的色泽也不同。有人烧红烧肉时喜欢加冰糖(砂糖重结晶产物),烧出的红烧肉色泽光亮,道理说不清楚,这有可能与药物的不同晶型产生不同的药效有点类似。建议平时烧红肉不要用市场上的肉香香精,而是用我们老祖宗积累的经验做红烧肉,加糖、黄酒、桂皮、生姜、八角等天然调味佐料。这些佐料中的化学物质与肉中成份产生复杂的化学反应,除酯化反应、美拉德反应、糖焦化反应(见下),其他反应目前不清楚,可能有全新的化学反应,值得研究。美拉德反应是食品化学研究的重要领域,每年有很多论文发表,目前还应用于疾病预防,例如有人研究出有利于糖尿病和慢性肾病患者食用的烤牛排的烹饪条件。我国科学家还用色谱和质谱技术研究北京烤鸭香味的指纹图谱,指纹图谱中的化合物多数是美拉德反应的产物。今后,如果把我国的高级厨师、民间烧菜高手做的红烧肉进行化学分析,做成指纹图谱数据库,并与他们烧制的配方和工艺过程相关联,进行大数据分析,产生出系列红烧肉烹饪工艺,家家户户都可以烧出适合自己口味的红烧肉。借此机会,我介绍一种不用酱油上色而是用糖饴(焦糖)上色红烧肉做法。油中热后,加入白糖,小火加热,搅拌,糖融化微焦并冒小泡时,加入处理好的五花肉翻炒2分钟左右,加入黄酒和上述佐料,倒入砂锅,再加黄酒(量至将肉刚好浸泡),小火烧15分钟,加盐,再烧10~15分钟左右。糖饴或焦糖的制作是一个更复杂的化学反应过程,主要涉及两类反应,一种是上述介绍的美拉德反应,另一类是糖加热的焦糖化反应,即在相当高的高温下(大约200℃)使碳水化合物产生醛类,然后缩合成有色成份。糖饴上色红烧肉的第一步,即糖在植物油中高温加热融化的过程是糖焦化反应,加入肉和其他佐料开始烧红烧肉时主要是美拉德反应,最后大火收汁时,焦化反应和美拉德反应同时发生。实际上,我们平时吃的酱油、醋、啤酒、可口可乐等佐料和饮料的颜色全靠焦糖着色。如果学会了用纯糖着色法烧红烧肉,就不用酱油了。要制作好的焦糖十分困难,因此,焦糖在也可称为高科技产品,每个公司的生产工艺均严格保密。可口可乐最关键的成份是一种耐酸焦糖,这是可口可乐之所以能风行全世界、在国际市场独占鳌头的主要原因,至今没人能破解这种耐酸焦糖的制作工艺。在这里,我也介绍一种制作焦糖的方法,用这种方法制作的焦糖特别适合于做红烧肉。我13岁时,曾跟一位老家的糕饼师傅学做炒米糖(米花糖),关于米花糖的做法我曾专门写过文章,这里不作介绍。做炒米糖的关键一步是熬糖,即制作糖饴。将大块的麦芽糖敲成细块,放入铁锅中融化。为防止糖熬焦,要加少量的水和猪油,与麦芽糖一起熬,火先大后小。因麦芽糖的甜度有限,熬糖时加一些白糖一起熬。掌握火候和熬到什么程度是关键,熬糖不到火候,粘度不够,炒米粘不起来,熬过头了,糖熬焦,炒米糖吃口就不好。这里有一个判断的标准,拿起搅拌的锅铲,如果糖汁一片一片往下落,就好了。这时,加入炒米搅拌,铲出后放在大一点的刀砧板上,压成方块。等糖块凉后,先切成条,再把条切成片,炒米糖就做好了。还有很多糖饴粘在铁锅上,千万不要洗掉,而是加入少量的水,继续加热熬,熬到略为粘稠时,倒在容器中备用。这是上等糖饴,下次做红烧肉时,不用上面介绍方面现做焦糖上色,而是直接加适量的这种糖饴,然后加其他需要的佐料,烧出的红烧肉味道一流。这种做糖饴的方法也发生了糖焦化反应和美拉德反应,熬麦芽糖时,肯定发生糖焦化反应,加入的猪油中含有少量的蛋白质或多肽,与麦芽糖发生美拉德反应。有很多高手烧的红烧肉味道很好,除其他佐料外,关键是他们加糖适量、火候处理得当,产生的美拉德反应和糖焦化反应的产物就好,无意中成了美拉德反应和糖焦化反应的高手。当然,做红烧鱼、红烧鸡、焙烤饼干面包、烤红薯、甚至炮制中药,只要是氨基酸和糖加热的过程,都会发生美拉德反应,也会发生糖焦化反应。美拉德反应和糖焦化反应也就成为食品化学和香料化学中的著名反应。本文介绍这些反应,希望今后大家烧红烧肉、红烧鱼时能有的放矢地利用这些反应。(2015年11月29日于杭州火车站,11月30日修改于上海药物研究所)
聚合酶链式反应(Polymerase Chain Reaction,PCR)是体外酶促合成特异DNA片段的一种方法,为最常用的分子生物学技术之一。典型的PCR由(1)高温变性模板;(2)引物与模板退火;(3)引物沿模板延伸三步反应组成一个循环,通过多次循环反应,使目的DNA得以迅速扩增。其主要步骤是:将待扩增的模板DNA置高温下(通常为93℃-94℃)使其变性解成单链;人工合成的两个寡核苷酸引物在其合适的复性温度下分别与目的基因两侧的两条单链互补结合,两个引物在模板上结合的位置决定了扩增片段的长短;耐热的DNA聚合酶(Taq酶)在72℃将单核苷酸从引物的3’端开始掺入,以目的基因为模板从5’→3’方向延伸,合成DNA的新互补链。 PCR能快速特异扩增任何已知目的基因或DNA片段,并能轻易在皮克(pg)水平起始DNA混合物中的目的基因扩增达到纳克、微克、毫克级的特异性DNA片段。因此,PCR技术一经问世就被迅速而广泛地用于分子生物学的各个领域。它不仅可以用于基因的分离、克隆和核苷酸序列分析,还可以用于突变体和重组体的构建,基因表达调控的研究,基因多态性的分析,遗传病和传染病的诊断,肿瘤机制的探索,法医鉴定等诸多方面。通常,PCR在分子克隆和DNA分析中有着以下多种用途:(1) 生成双链DNA中的特异序列作为探针;(2) 由少量mRNA生成 cDNA文库;(3) 从cDNA中克隆某些基因;(4) 生成大量DNA以进行序列测定;(5) 突变的分析;(6) 染色体步移;(7) RAPD、AFLP、RFLP等DNA多态性分析等。一、试剂准备1. DNA模版2.对应目的基因的特异引物3.10×PCR Buffer 4.2mM dNTPmix:含dATP、dCTP、dGTP、dTTP各2mM5.Taq酶二、操作步骤 1.在冰浴中,按以下次序将各成分加入一无菌0.5ml离心管中。 10×PCR buffer 5 μl dNTP mix (2mM) 4 μl 引物1(10pM) 2 μl 引物2(10pM) 2 μl Taq酶 (2U/μl) 1 μl DNA模板(50ng-1μg/μl) 1 μl 加ddH2O至 50 μl 视PCR仪有无热盖,不加或添加石蜡油。2. 调整好反应程序。将上述混合液稍加离心,立即置PCR仪上,执行扩增。一般:在93℃预变性3-5min,进入循环扩增阶段:93℃ 40s → 58℃ 30s → 72℃ 60s,循环30-35次,最后在72℃ 保温7min。3. 结束反应,PCR产物放置于4℃待电泳检测或-20℃长期保存。4.PCR的电泳检测:如在反应管中加有石蜡油,需用100μl氯仿进行抽提反应混合液,以除去石蜡油;否则,直接取5-10μl电泳检测。三、PCR反应体系的组成与反应条件的优化 PCR反应体系由反应缓冲液(10×PCR Buffer)、脱氧核苷三磷酸底物(dNTPmix)、耐热DNA聚合酶(Taq酶)、寡聚核苷酸引物(Primer1,Primer2)、靶序列(DNA模板)五部分组成。各个组份都能影响PCR结果的好坏。1. 反应缓冲液:一般随Taq DNA聚合酶供应。标准缓冲液含:50mM KCl,10mM Tris-HCl(pH8.3室温),1.5mM MgCl2。Mg2+的浓度对反应的特异性及产量有着显著影响。浓度过高,使反应特异性降低;浓度过低,使产物减少。在各种单核苷酸浓度为200μM时,Mg2+为1.5mM较合适。若样品中含EDTA或其它螯合物,可适当增加Mg2+的浓度。在高浓度DNA及dNTP条件下进行反应时,也必须相应调节Mg2+的浓度。据经验,一般以1.5-2mM(终浓度)较好。2. dNTP :高浓度dNTP易产生错误掺入,过高则可能不扩增;但浓度过低,将降低反应产物的产量。PCR中常用终浓度为50-400μM的dNTP。四种脱氧三磷酸核苷酸的浓度应相同,如果其中任何一种的浓度明显不同于其它几种时(偏高或偏低),就会诱发聚合酶的错误掺入作用,降低合成速度,过早终止延伸反应。此外,dNTP能与Mg2+结合,使游离的Mg2+浓度降低。因此,dNTP的浓度直接影响到反应中起重要作用的Mg2+浓度。3. Taq DNA聚合酶酶:在100μl反应体系中,一般加入2-4U的酶量,足以达到每min延伸1000-4000个核苷酸的掺入速度。酶量过多将导致产生非特异性产物。但是,不同的公司或不同批次的产品常有很大的差异,由于酶的浓度对PCR反应影响极大,因此应当作预试验或使用厂家推荐的浓度。当降低反应体积时(如20μl或50μl),一般酶的用量仍不小于2U,否则反应效率将降低。4. 引物:引物是决定PCR结果的关键,引物设计在PCR反应中极为重要。要保证PCR反应能准确、特异、有效地对模板DNA进行扩增,通常引物设计要遵循以下几条原则:⑴ 引物的长度以15-30bp为宜,一般(G+C)的含量在45-55%,Tm值高于55℃。应尽量避免数个嘌呤或嘧啶的连续排列,碱基的分布应表现出是随机的。⑵ 引物的3’端不应与引物内部有互补,避免引物内部形成二级结构,两个引物在3’端不应出现同源性,以免形成引物二聚体。3’端末位碱基在很大程度上影响着Taq酶的延伸效率。两条引物间配对碱基数少于5个,引物自身配对若形成茎环结构,茎的碱基对数不能超过3个由于影响引物设计的因素比较多,现常常利用计算机辅助设计。⑶ 人工合成的寡聚核苷酸引物需经PAGE或离子交换HPLC进行纯化。⑷ 引物浓度不宜偏高,浓度过高有两个弊端:一是容易形成引物二聚体(primer-dimer),二是当扩增微量靶序列并且起始材料又比较粗时,容易产生非特异性产物。一般说来,用低浓度引物不仅经济,而且反应特异性也较好。一般用0.25-0.5pM/μl较好。⑸ 引物一般用TE配制成较高浓度的母液(约100μM),保存于-20℃。使用前取出其中一部分用ddH2O配制成10μM或20μM的工作液。5. 模板:PCR对模板的要求不高,单、双链DNA均可作为PCR的样品。虽然PCR可以用极微量的样品(甚至是来自单一细胞的DNA)作为摸板,但为了保证反应的特异性,一般还宜用μg水平的基因组DNA或104拷贝的待扩增片段作为起始材料。原材料可以是粗制品,某些材料甚至仅需用溶剂一步提取之后即可用于扩增,但混有任何蛋白酶、核酸酶、Taq DNA聚合酶抑制剂以及能结合DNA的蛋白,将可能干扰PCR反应。6. PCR循环加快,即相对减少变性、复性、延伸的时间,可增加产物的特异性。四、注意事项1.PCR反应应该在一个没有DNA污染的干净环境中进行。最好设立一个专用的PCR实验室。2.纯化模板所选用的方法对污染的风险有极大影响。一般而言,只要能够得到可靠的结果,纯化的方法越简单越好。3.所有试剂都应该没有核酸和核酸酶的污染。操作过程中均应戴手套。4.PCR试剂配制应使用最高质量的新鲜双蒸水,采用0.22μm滤膜过滤除菌或高压灭菌。5.试剂都应该以大体积配制,试验一下是否满意,然后分装成仅够一次使用的量储存,从而确保实验与实验之间的连续性。6.试剂或样品准备过程中都要使用一次性灭菌的塑料瓶和管子,玻璃器皿应洗涤干净并高压灭菌。7.PCR的样品应在冰浴上化开,并且要充分混匀。
讨论:除了还原糖、氨基酸、HVP、酵母抽提物,还有哪些是通过美拉德反应产生肉风味的主要原料?常用的还原糖和氨基酸有哪些???美拉德反应的通常温度是在110度-120度之间??
想在实验室搭建一套水煤气发生装置,但先前没有人做过类似方向,所以对实验所需装置不是很清楚。 目前是想做催化甲烷和水蒸气制水煤气,实验室目前已有气-固反应床,需要接通气体的装置,但现在不清楚该选用什么配件如何连接。请问有没有做过类似方向的朋友能给下建议。如果能给一个完整的反应装置图,就更加感激不尽了[img]http://simg.instrument.com.cn/bbs/images/default/em09505.gif[/img]
最近做了实验,要对CaO和煤反应后固相产物进行分析,一次实验是2.5gCaO和2.5g煤反应,一次实验是2.5CaO和5g煤反应,请问有什么方法能分析出固相产物的不同吗?谢谢
聚合酶链反应(PC)技术的发展和应用作者:antony第一节 概述 聚合酶链反应或多聚酶链反应(Polymerase Chain Reaction, PCR),又称无细胞克隆技术(“free bacteria”cloning technique),是一种对特定的DNA片段在体外进行快速扩增的新方法。该方法一改传统分子克隆技术的模式,不通过活细胞,操作简便,在数小时内可使几个拷贝的模板序列甚至一个DNA分子扩增107~108倍,大大提高了DNA的得率。因此,现已广泛应用到分子生物学研究的各个领域。 PCR技术最早由美国Cetus公司人类遗传研究室Kary Mullis及同事于1985年发现并研制成功的;最早的应用报道是Saiki等1985年将PCR技术应用于β-珠蛋白基因扩增和镰刀状红细胞贫血的产前诊断。随后使用了1976年Chien等分离的热稳定性Taq DNA聚合酶,使PCR操作大为简化,并使PCR自动化成为可能;1987年Kary Mullis等完成了自动化操作装置,使PCR技术进行实用阶段。 国内复旦大学1988年起开始研制耐热性多聚酶,军事医学科学院马立人教授等在1989年研制成功了PCR自动装置,并且不断推陈出新,最近研制的PTC-51A/B型DNA热
最近做了实验,要对CaO和煤反应后固相产物进行分析,一次实验是2.5gCaO和2.5g煤反应,一次实验是2.5CaO和5g煤反应,请问有什么方法能分析出固相产物的不同吗?谢谢
想研究碘与腐殖质、酶的反应,可以用哪些仪器呢
最近做了实验,要对CaO和煤反应后固相产物进行分析,一次实验是2.5gCaO和2.5g煤反应,一次实验是2.5CaO和5g煤反应,请问有什么方法能分析出固相产物的不同吗?谢谢!
测定土样蔗糖酶时要在三角瓶中放置24小时,然后要6000rpm离心,这样就必须从三角瓶中移到离心管里,不用水冲洗的前提下有没有办法将三角瓶里的反应溶液倒干净,有没有必要倒干净,如果没法倒干净,直接将土样和测定的反应液放置在离心管里放置24小时,然后直接离心可以吗?
苯并吡喃酮作为一个非常重要的结构内核单元,广泛地存在于很多天然产物中,并且是一些具有生物活性的化合物的特定前导化合物。此外该类化合物也因其具有一些光致变色效应和热致变色性质而受到广泛关注。 生物催化作为一种绿色高效的现代有机合成技术,在医药、能源、材料等领域显示出巨大的潜力。酶作为一种高效,环境友好的催化剂,已经成为传统有机金属和小分子催化剂的有效补充,尤其是酶的非专一性在有机合成方面的应用备受研究者的青睐。近些年,已有一系列关于水解酶催化非专一性的C-C键以及C-杂原子键的形成反应的报道,但是对于单酶催化的多步串联反应的报道较少。基于此,我们设计和发展基于水解酶催化非专一性的domino反应,发现枯草杆菌α-淀粉酶可以有效地催化水杨醛和丁烯酮发生oxa-Michael/aldol缩合反应合成2H-苯并吡喃酮类化合物。2H-苯并吡喃酮的生物合成在25 mL烧瓶中加入水杨醛(2 mmol),不饱和醛或酮(10 mmol),50 mg 枯草杆菌α-淀粉酶(BSA),再加入500μL去离子水和4.5 mL DMSO,将其置于50℃恒温摇床中震荡反应(200 rpm)。TLC跟踪检测反应(紫外灯下观测),反应完全后,加入20 mL蒸馏水停止反应,然后用乙酸乙酯萃取(3×10 mL),有机相用无水硫酸钠干燥后,经减压蒸馏除去溶剂,经柱层析分离得目标产物(硅胶:300-400目,流动相:石油醚[font='Times
[B][center]聚合酶链反应(PCR)技术的发展和应用(一) [/center][/B] 第一节 概述 聚合酶链反应或多聚酶链反应(Polymerase Chain Reaction, PCR),又称无细胞克隆技术(“free bacteria”cloning technique),是一种对特定的DNA片段在体外进行快速扩增的新方法。该方法一改传统分子克隆技术的模式,不通过活细胞,操作简便,在数小时内可使几个拷贝的模板序列甚至一个DNA分子扩增107~108倍,大大提高了DNA的得率。因此,现已广泛应用到分子生物学研究的各个领域。 PCR技术最早由美国Cetus公司人类遗传研究室Kary Mullis及同事于1985年发现并研制成功的;最早的应用报道是Saiki等1985年将PCR技术应用于β-珠蛋白基因扩增和镰刀状红细胞贫血的产前诊断。随后使用了1976年Chien等分离的热稳定性Taq DNA聚合酶,使PCR操作大为简化,并使PCR自动化成为可能;1987年Kary Mullis等完成了自动化操作装置,使PCR技术进行实用阶段。 国内复旦大学1988年起开始研制耐热性多聚酶,军事医学科学院马立人教授等在1989年研制成功了PCR自动装置,并且不断推陈出新,最近研制的PTC-51A/B型DNA热循环仪体积小,造型美观,价格适宜,操作简单,尤为适宜国内应用。 PCR发明不到10年,却已获得广泛应用。目前,每年都有上千篇文章 发表。1991年,期刊“PCR方法与应用”(PCr Methods and Application)在美国创刊,使有关学者有了自己的论坛和参考的专业期刊。PCR技术作为一种方法学革命,必将大大推动分子生物学各有关学科的研究,使其达到一个新的高度。 1993年度诺贝尔化学将已于10月13日揭晓,Kary Mullis因发明了“聚合酶链式反应”而获得此殊荣。现在世界各地都在使用PCR检测病人血液中的微量遗传物质,这一成就为精确诊断艾滋病及其它病症铺平了道路。瑞典皇家科学院说:“PCR方法已经广泛应用于生物医学中。该方法同DNA测序法结合起来很可能将成为研究动植物分类学的一种革新工具。”一名加拿大籍英国科学家Michael Smith因开创了“寡核苷酸基因定点诱变”的方法而与Mullis同享此荣。
《中国药典》二部对镁盐的鉴别罗列了两种方法:其一:取供试品溶液,滴加氨试液即产生白色沉淀,加氯化铵试液,沉淀溶解,再加磷酸氢二钠试液1滴,振摇,即生成白色沉淀,沉淀不溶于氨试液。对于这个方法做含有次硝酸铋的碳酸镁鉴别时,专属性不好,在加入氯化铵试液时,沉底不溶解,据我了解应该是次硝酸铋的干扰原因,请问哪位高手知道在次鉴别的前处理上将铋盐掩蔽而不影响镁盐反应的方法?多谢了
用蛋白酶反应的反应液里面的蛋白怎么去除才能做[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]?用有机溶剂除不干净,用三氯乙酸还得除盐,用超滤膜行吗?有没有推荐的?
[color=#444444]最近在做4cl家族基因的重组蛋白和五种底物酚酸反应的实验,反应体系中存在0.6mM CoA、5mM ATP、6mM MgCl2、0.2M Tris-Hcl、0.2mM 酚酸底物以及浓度不明的重组蛋白。配制了7种不同PH的Tris-Hcl缓冲液(PH=4、5、6、7.1、7.5、8.1、8.9),体系反应10分钟后,沸水煮沸10分钟失活,现使用对香豆酸和酶反应,反应后经12000RAM离心2min后用0.22um过滤头过滤,进样10ul,使用的是XDB-C18的柱子,waters2695的色谱仪,流动相为A1%乙酸和B纯乙腈,洗脱梯度0~5min A:B=95:5,5-35min A:B=25:75,36-5,5min A:B=95:5,56-65min A:B=5:95 进样后发现,除了PH=7.1的样品外其他的样品都没有底物(对-香豆酸)峰,并且所有样品都没有产物(对-香豆酸辅酶A)峰,能请教一下各位大神,怎么解决这个问题[/color][color=#444444] 另外还有一个问题,五种酚酸跑混标的时候,芥子酸和阿魏酸总是分不开,大神有没有什么好的方法将他们分开(尽量不使用缓冲盐体系作为流动相),多谢各位了[/color]
浓硫酸和金属镁在常温下怎么反应?
最近在做4cl家族基因的重组蛋白和五种底物酚酸反应的实验,反应体系中存在0.6mM CoA、5mM ATP、6mM MgCl2、0.2M Tris-Hcl、0.2mM 酚酸底物以及浓度不明的重组蛋白。配制了7种不同PH的Tris-Hcl缓冲液(PH=4、5、6、7.1、7.5、8.1、8.9),体系反应10分钟后,沸水煮沸10分钟失活,现使用对香豆酸和酶反应,反应后经12000RAM离心2min后用0.22um过滤头过滤,进样10ul,使用的是XDB-C18的柱子,waters2695的色谱仪,流动相为A1%乙酸和B纯乙腈,洗脱梯度0~5min A:B=95:5,5-35min A:B=25:75,36-5,5min A:B=95:5,56-65min A:B=5:95 进样后发现,除了PH=7.1的样品外其他的样品都没有底物(对-香豆酸)峰,并且所有样品都没有产物(对-香豆酸辅酶A)峰,能请教一下各位大神,怎么解决这个问题 另外还有一个问题,五种酚酸跑混标的时候,芥子酸和阿魏酸总是分不开,大神有没有什么好的方法将他们分开(尽量不使用缓冲液体系作为流动相),多谢各位了
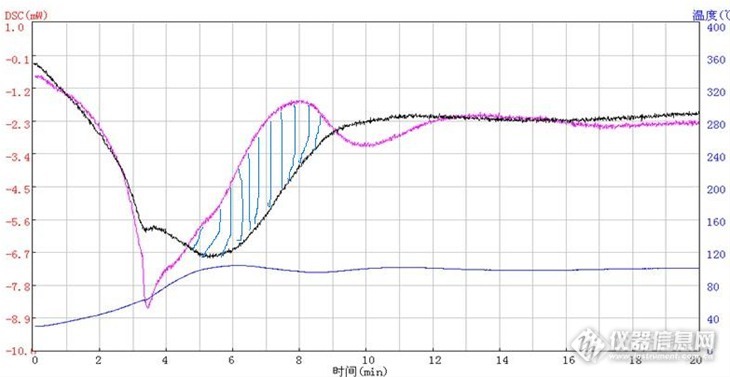
看到了南京大展的活动奖品,就忍不住赶紧来参赛了 \(≧▽≦)/。正好手上有一台南京大展的差示扫描量热仪DSC-100,目前也正在使用这台仪器做实验,今天就把我做的实验过程发上来,和大家一起讨论讨论 O(∩_∩)O。我们实验室的差热托盘及传感器是铂铑合金,灵敏度还是非常好的。这个实验之前在论坛里也和大家讨论过,就是用DSC测定磷钼酸与煤油反应放出的热量。听上去很简单吧、、But!!这实验可耗了我不少精力!!_ 最难解决的一个问题就是煤油与磷钼酸常温下一接触便反应,因此必须在将这两个物质放进炉子之前隔离。这点是最令人头疼的啊!!_ 但最终,功夫不负有心人,还是想到了一个办法,考虑到石蜡与煤油结构相似,石蜡是固体,可以用它将两个物质隔离开!石蜡的熔点在50~60℃左右,只要加热到石蜡熔化的温度,煤油就可以掉下去与磷钼酸反应啦(刚开始是考虑石蜡与煤油可以相似相溶,试了一下发现,需要的时间太长,而且石蜡与煤油相溶之后的流动性会变的非常差,最后还是用加热的方法了)。这是个很大的突破啊!!\(≧▽≦)/ 找到了隔离的物质,下面就好办啦~~(*^__^*) …下面先来隆重介绍下这个实验所用的铝坩埚,当当当当::::::图1 http://ng1.17img.cn/bbsfiles/images/2013/09/201309071627_462982_2692547_3.jpg图2 http://ng1.17img.cn/bbsfiles/images/2013/09/201309071627_462981_2692547_3.jpg图3 http://ng1.17img.cn/bbsfiles/images/2013/09/201309071628_462983_2692547_3.jpg图4 http://ng1.17img.cn/bbsfiles/images/2013/09/201309071628_462985_2692547_3.jpg图5 http://ng1.17img.cn/bbsfiles/images/2013/09/201309071628_462984_2692547_3.jpg石蜡与煤油性质相似,石蜡也可能会跟磷钼酸反应,因此,石蜡跟煤油一样,在放进炉子之前也不能碰到磷钼酸,这样我就想到了做如图1、2所示的坩埚,下层大坩埚放磷钼酸口部收紧刚好能夹紧上层小坩埚,上层小坩埚底部中央开孔,再用石蜡封住孔,然后放上煤油。\(≧▽≦)/很聪明吧!多不容易啊~~可是实际测量过程中发现,下面的大坩埚口会松掉,导致上层坩埚底部的石蜡与磷钼酸直接接触,还有一个问题就是由于上层坩埚较小,石蜡熔化后任粘在孔上,煤油掉不下去无法与磷钼酸反应。于是把坩埚改进了一下,如图3、4、5,上层为大坩埚,底部开孔封蜡,然后放上煤油,下层坩埚放磷钼酸。问题是接踵而至啊~~~刚解决了坩埚的问题,下面的问题更复杂呢~~~~~~俗话说,有利就有弊,石蜡隔离了两个物质,同时也带入了新的误差因素。石蜡会与磷钼酸反应的话,那么他们也会放出热量,最终测得的放热热焓要减去石蜡与磷钼酸反应的热焓。另外石蜡、煤油、磷钼酸加热时都会吸热,一正一负会相互抵消╮(╯0╰)╭。这是要麻烦死人的节凑嘛(⊙_⊙)!?为了消除各组分的吸热,为了能更准确的算出煤油与磷钼酸的放热热焓,我制定了这样的方案,一组实验包括:a.石蜡+煤油+磷钼酸用图5所示的坩埚测量(上层坩埚打孔);f.石蜡+煤油+磷钼酸用上层坩埚底部不打孔,其他与图5所示一样的双层坩埚测量(也就是说,让煤油、石蜡不接触磷钼酸,这样可以消除三个物质的吸热总量);c.石蜡+磷钼酸反应测[font
怎么除去美拉德反应样品中的葡萄糖?溶剂是丙二醇,葡萄糖是反应物,目标物是挥发性成分,需要进气相检测,但是反应物带来的本底好高,估计是受葡萄糖影响.之前用戊烷萃取过反应物,基线很好,但是部分产物萃取不出来,不能满足分析需要. 所以来这里诚心向各位请教,怎么除去样品中的糖?有没有沉淀离心法合适的试剂?或者其他处理方法







 我要推广仪器
我要推广仪器
 下载APP
下载APP