青龙衣green walnut husks来源于胡桃科胡桃属植物胡桃和胡桃楸的未成熟果实的干燥外果皮。胡桃醌是青龙衣中的萘醌类化合物,也是主要活性成分[1]。现代药理研究表明,胡桃醌具有抗炎、抗菌、抗肿瘤等作用[2-5],对多种肿瘤细胞增殖均有抑制作用。目前,已证实胡桃醌能抑制宫颈癌细胞生长,并诱导其凋亡、抑制细胞迁移、侵袭[6-7]。其对肝癌HepG2细胞的体内外抑制活性显著,能够上调死亡受体5(death receptor 5,DR5)表达,通过ROS介导的p53信号通路激活,促进自噬体形成,诱导细胞的凋亡与自噬[8]。胡桃醌对人乳腺癌MCF-7细胞抑制生长效果明显,与时间和浓度呈正相关,同时使Bcl-2相关X蛋白/B淋巴细胞瘤-2(Bcl-2 associated X protein/B-cell lymphoma-2,Bax/Bcl-2)比值升高,半胱氨酸天冬氨酸蛋白酶-3(cystein- asparate protease-3,Caspase-3)、Caspase-9被激活,诱导细胞凋亡[9]。但胡桃醌水溶性差,易升华,能随水蒸汽挥发,长期存放易发生氧化分解,限制了其在新药开发和在临床上的应用[10],因此,针对其药理活性及潜在应用,设计一种可有效提高胡桃醌稳定性的递药体系具有重要意义。 两亲性嵌段共聚物是在自组装过程中将疏水性药物包覆或键合在聚合物中形成的载药纳米胶束,其能够弥补传统药物水溶性差、吸收率低等不足,可提高药物生物利用度,实现靶向控制释放,在抗癌药物递送中被广泛应用[11]。白及多糖(Bletilla striata polysaccharide,BSP)是从兰科白及属植物白及Bletilla striata (Thunb.) Reichb. f.的干燥块茎中提取得到的一类水溶性多糖,作为天然高分子材料,具有结构稳定、生物可降解、生物安全性高、易于修饰改造等特点,逐渐成为一种纳米药物递送系统的新型优良载体材料[12]。维生素E琥珀酸酯(vitamin E succinate,VES)是维生素E的类似物,因具有较长的脂肪链而疏水性较强,将其和白及多糖连接可提高包载药物的稳定性。VES还能够抑制肿瘤细胞生长和诱导肿瘤细胞凋亡,且只对肿瘤细胞有抑制作用,对正常的组织细胞无任何不良反应,因此VES具有药物和载体的双重作用[13-14],在递送药物的同时达到辅助治疗的效果。 本实验以白及多糖为亲水端,VES为疏水端,合成两亲性嵌段共聚物BSP-VES,将其作为载体制备胡桃醌载药胶束(Jug/BSP-VES),同时考察制备过程中各因素对包封率和载药量的影响,采用星点设计-效应面法(central composite design-response surface methodology,CCD-RSM)优化Jug/BSP-VES胶束的处方和工艺,并进行质量评价,为传统中药青龙衣及其活性成分胡桃醌的开发及临床应用提供参考。 1 仪器与材料 1.1 仪器 Agilent 1260 Series型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],美国安捷伦有限公司;DF-101S型集热式恒温加热磁力搅拌器,上海秋佐科学仪器有限公司;KQ-200KDB型超声波清洗器,昆山市超声仪器有限公司;UV-765型紫外-可见分光光度计,上海精密科学仪器有限公司;Advantage型台式托盘冻干机,美国VirTis公司;80-2型电动离心机,上海浦东物理光学仪器厂;Zetasizer Nano ZSE型纳米粒度电位仪,英国马尔文公司;FTIR-650型傅里叶变换红外光谱仪,天津港东科技股份有限公司;970CRT型荧光分光光度计,北京恒奥德仪器有限公司;Hula Dancer Digital型涡旋混合器,德国IKA公司;Talos F200S G2型透射电子显微镜(TEM),赛默飞仪器公司。 1.2 试药 胡桃醌原料药(批号A2007171,质量分数≥97%)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC)、4-二甲氨基吡啶(DMAP),上海阿拉丁试剂有限公司;白及多糖,批号GH210721,西安国豪生物科技有限公司;胡桃醌对照品,批号RFS-H07511804026,质量分数>98%,成都瑞芬思生物科技有限公司;VES,批号VS1210200734,西安海斯夫生物科技有限公司;芘,分析纯,上海九鼎化学有限公司。 2 方法与结果 2.1 BSP-VES聚合物的合成 称取3.2 g BSP超声溶解于30 mL DMSO中。另称取适量VES、DMAP和EDC(nVES∶nDMAP∶nEDC=1∶1∶1.2)溶于DMSO后磁力搅拌活化1 h,BSP溶液缓慢滴入,密封圆底烧瓶,38 ℃水浴搅拌下反应48 h,室温冷却后移至透析袋(截留相对分子质量3 500)中用纯化水透析2 d以除去未反应试剂。将溶液3 500 r/min离心(离心半径10 cm)15 min取上清液,?20 ℃冰箱中预冻,随后进行冷冻干燥,得到棕色絮状疏松固体,置于4 ℃冰箱中冷藏备用,反应式见图1。 图片 2.2 BSP-VES的表征及结果 2.2.1 核磁共振氢谱(1H-NMR)检测 以D2O为溶剂,对BSP-VES合成产物进行1H-NMR分析。结果如图2所示,δ 3.0~4.0处宽峰为白及多糖上甘露糖和葡萄糖单元中的亚甲基和次甲基(CH2-O和CH-O)的质子峰,δ 0.8~1.0附近为VES中甲基(e)、亚甲基信号峰,δ 5.31处为白及多糖(1,6)糖苷键(a)的质子化学位移。以上结果表明合成产物为BSP-VES[15]。 图片 2.2.2 红外光谱(IR)检测 采用IR法分别对BSP、VES、BSP-VES进行表征,红外扫描范围为4 000~500 cm?1,结果如图3所示。BSP的结果图(图3-a)中,3 384.56、2 921.63 cm?1为O-H和C-H的伸缩振动峰,1 149.37、1 076.08、1 025.94 cm?1为吡喃糖苷构型的特征峰。VES的结果图(图3-b)中,2 923.56 cm?1为-CH2、-CH的伸缩振动峰,1 749.12、1 710.55 cm?1为羧基和酯基中C=O伸缩振动峰,1 373.07、1 157.08 cm?1为-CH3和C-O的伸缩振动峰。BSP-VES的结果图(图3-c),其中2 921.63 cm?1处的C-H伸缩振动峰增强,说明有VES中大量-CH2、-CH3的引入,1 739.48 cm?1为酯基中C=O伸缩振动峰,1 567.84 cm?1为VES中苯环骨架振动峰,揭示了VES的引入[16]。 图片 2.3 Jug/BSP-VES胶束的制备 采用溶剂挥发法制备Jug/BSP-VES胶束[17]。称取20 mg的BSP-VES于15 mL水中,称取2 mg胡桃醌溶于3 mL无水乙醇中,在搅拌下将含药溶液滴加至水相中,在30 ℃下搅拌6 h,有机溶剂挥发完全后即得Jug/BSP-VES胶束溶液。预冻后,置于冻干机中,取出即得冻干粉。 2.4 Jug/BSP-VES中胡桃醌含量测定方法 2.4.1 色谱条件 色谱柱为依利特Kromasil(250 mm×4.6 mm,5 μm);流动相为甲醇-水(70∶30);检测波长248 nm;柱温25 ℃;体积流量1.0 mL/min;进样量10 μL。 2.4.2 溶液的配制 (1)对照品溶液的配制:精密称取胡桃醌对照品5.0 mg,置于25 mL量瓶中,甲醇溶解并定容,得质量浓度为200 μg/mL的对照品储备液。 (2)供试品溶液的配制:精密吸取Jug/BSP-VES胶束溶液0.5 mL至10 mL量瓶中,甲醇破乳并定容至刻度,摇匀,即得Jug/BSP-VES供试品溶液。空白胶束供试品溶液同法操作。 2.4.3 专属性考察 分别取适量空白胶束供试液、适当浓度的胡桃醌对照品溶液及Jug/BSP-VES供试品溶液各10 μL,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],按“2.4.1”项下色谱条件测定,记录色谱图。结果见图4,空白胶束在胡桃醌处无干扰,专属性良好。 图片 2.4.4 线性关系考察 取“2.4.2”项下对照品溶液适量,加甲醇稀释,得到系列质量浓度为1、5、10、30、50、70、100 μg/mL的对照品溶液,按“2.4.1”项下色谱条件进样测定,记录峰面积,以峰面积(Y)对质量浓度(X)进行线性方程拟合,得回归方程为Y=44.786 X-38.423,r=0.999 8,结果表明胡桃醌在1~100 μg/mL线性关系良好。 2.4.5 精密度试验 取“2.4.4”项下低、中、高3个质量浓度(分别为5、30、70 μg/mL)胡桃醌对照品溶液,同1 d内各质量浓度分别进样5次,计算日内精密度;各质量浓度连续进样5 d,计算日间精密度。日内与日间精密度RSD均小于2.0%,表明仪器的精密度良好。 2.4.6 稳定性试验 精密吸取同一供试品溶液在0、2、4、8、12、24 h下,按照“2.4.1”项下色谱条件进行测定,结果峰面积的RSD值为0.596%,表明供试品溶液在24 h内稳定性良好。 2.4.7 重复性试验 取同一批Jug/BSP-VES 6份,按“2.4.2”项方法制备供试品溶液,按照“2.4.1”项下色谱条件进行测定,计算胡桃醌质量浓度的RSD值为1.03%,表明测定方法的重复性良好。 2.4.8 加样回收率试验 精密量取200 μg/mL胡桃醌对照品溶液0.25、1.50、3.50 mL各3份于10 mL量瓶中,加入BSP-VES聚合物,用甲醇定容,分别得到胡桃醌质量浓度为5、30、70 μg/mL的溶液,按“2.4.1”项下色谱条件测定胡桃醌的含量,测得加样回收率均在99%~102%,RSD均小于2.0%,表明检测结果准确可靠。 2.5 胡桃醌包封率、载药量的测定 采用离心法进行聚合物胶束药物包封率和载药量的测定[18]。精密吸取Jug/BSP-VES胶束溶液1 mL至1.5 mL离心管中,3 000 r/min离心(离心半径8 cm)10 min,除去游离药物,吸取0.5 mL上清液,甲醇破乳并定容至刻度,摇匀,按“2.4.1”项下色谱条件进样分析。另取Jug/BSP-VES胶束溶液0.5 mL至10 mL量瓶中,甲醇破乳并定容至刻度,摇匀,按“2.4.1”项下色谱条件进样分析。将所得峰面积带入线性方程计算胡桃醌的包封率和载药量。 包封率=W胶束中药物量/W总药量 载药量=W胶束中药物量/W胶束质量 2.6 单因素考察 2.6.1 有机溶剂种类考察 固定其他条件不变,即有机溶剂用量为3 mL,挥发时间为6 h,制备温度为30 ℃,载药比为10∶1,水相用量为15 mL,分别加入有机溶剂氯仿、丙酮、甲醇、无水乙醇,考察不同有机溶剂种类对载药量和包封率的影响。结果(表1)显示,以无水乙醇为溶剂时,制备的胶束溶液包封率和载药量最高,因此,选择无水乙醇作为溶剂来制备Jug/BSP-VES胶束。 图片 2.6.2 有机溶剂用量考察 固定其他条件不变,即有机溶剂为无水乙醇,挥发时间为6 h,制备温度为30 ℃,载药比为10,水相用量为15 mL,加入一定量的BSP-VES和胡桃醌分别溶解于1、2、3、4、5 mL无水乙醇中,考察不同有机溶剂用量对载药量和包封率的影响。结果(表2)显示,当有机溶剂用量为3 mL时胡桃醌的载药量和包封率最高,因此,选择3 mL作为有机溶剂用量。 图片 2.6.3 挥发时间考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,制备温度为30 ℃,载药比为10,水相用量为15 mL,考察挥发时间在4、5、6、7、8 h时,不同挥发时间对载药量和包封率的影响。结果(表3)显示,当挥发时间为6 h时胡桃醌的载药量和包封率最高,因此,选择6 h作为挥发时间来制备Jug/BSP-VES胶束。 图片 2.6.4 制备温度考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,载药比为10,水相用量为15 mL,考察制备温度在25、30、35、40、45 ℃时,不同制备温度对载药量和包封率的影响。结果(表4)显示,随着制备温度的增加,胡桃醌的载药量与包封率先升高后降低,因此将25~35 ℃的制备温度作为待优化项进行CCD-RSM实验。 图片 2.6.5 载药比考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,制备温度为30 ℃,水相用量为15 mL,精密称取药物2 mg,加入不同质量的载体,即载药比分别为6、8、10、12、14时,考察不同载药比对载药量和包封率的影响。结果(表5)显示,随着载体量的增加,胡桃醌的包封率先升高后降低,因此将8、10、12的载药比作为待优化项进行CCD-RSM实验。 图片 2.6.6 水相用量考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,制备温度为30 ℃,载药比为10,考察水相用量在5、10、15、20、25 mL时,不同水相用量对载药量和包封率的影响。结果(表6)显示,随着水相用量的增加,胡桃醌的载药量与包封率先升高后降低,因此将10~20 mL的水相用量作为待优化项进行CCD-RSM实验。 图片 2.7 CCD-RSM优化处方 在单因素考察实验基础上,进一步采用CCD- RSM优化制剂工艺。选取载药比(X1)、水相体积(X2)、制备温度(X3)3个因素,每个因素设定5个水平(?1.682、?1、0、+1、+1.682)。以胡桃醌包封率(Y1)和胡桃醌载药量(Y2)为考察指标进行3因素,5水平的CCD-RSM实验,结果见表7。采用Design-Expert统计软件对表7数据进行统计处理,并获得Y1、Y2值对自变量X1、X2、X3的多元线性回归方程,各考察指标的2项式拟合方程如下Y1=90.010+0.165 9 X1+0.700 4 X2-0.1071 X3-0.656 4 X1X2-1.020 X1X3-0.516 1 X2X3-4.430 X12-3.520 X22-4.100 X32;Y2=6.430-0.408 3 X1+0.288 5 X2-0.210 6 X3+0.251 8 X1X2-0.380 1 X1X3-0.374 7 X2X3-0.004 7 X12-0.057 7 X22-0.111 9 X32。各方程的方差分析结果见表8,结果表明该模型与实际试验拟合程度良好,且各因素影响显著用该模型分析和预测胶束的制备工艺是合适的。 图片 图片 利用Design-Expert统计软件绘制自变量对因变量的效应面和等高线图,结果见图5。最终确定最佳条件范围得到的最优处方:BSP-VES与胡桃醌的投药量分别为20 mg和2 mg,水相用量15 mL,制备温度30 ℃。预测在此条件下制备Jug/BSP-VES的包封率和载药量分别为90.047%、6.559%。 图片 2.8 最优处方的验证试验 按最优处方平行制备3批Jug/BSP-VES胶束溶液,测定其中胡桃醌的包封率、载药量。胡桃醌的平均包封率为(88.44±1.24)%、RSD值为1.79%,胡桃醌平均载药量为(6.54±0.02)%、RSD值为1.90%,RSD值均<3%,表明模型预测可靠,工艺重现性较好。 2.9 Jug/BSP-VES胶束的表征 2.9.1 Jug/BSP-VES胶束溶液外观及形态观察 取制备好的Jug/BSP-VES溶液,观察外观及丁达尔现象;取适量Jug/BSP-VES溶液纯水稀释,滴加至专用铜网上,待风干后,通过透射电子显微镜(TEM)观察形态并拍照。结果如图6所示,Jug/BSP-VES胶束溶液为黄色澄清溶液,丁达尔效应明显;在TEM下观察到Jug/BSP-VES胶束呈类球形,分散均匀。 图片 2.9.2 BSP-VES临界聚集浓度(critical aggregation concentration,CAC)的测定 采用芘荧光探针法检测聚合物的CAC。配制质量浓度为1 mg/mL的芘溶液和1 mg/mL的BSP-VES母液。取9个西林瓶,各加入0.25 mL芘溶液,氮气吹干后各加入不同质量浓度的1 mL BSP-VES溶液。稀释后BSP-VES溶液的质量浓度分别为100.00、50.00、10.00、5.00、1.00、0.50、0.10、0.05、0.01 μg/mL。涡旋5 min后超声30 min,室温避光静置24 h。荧光分光光度计的激发波长为330 nm,测定各溶液中芘的荧光吸收,以373、384 nm处样品的荧光光度值之比(I373/I384)对质量浓度的对数作图,两条切线的交点为CAC值。结果如图7所示,当BSP-VES质量浓度较低时,I373/I384值较小,当BSP-VES质量浓度增大时,I373/I384值增大,取图中两直线相交处为BSP-VES的CAC值,经计算,CAC值为5.95 μg/mL。 图片 2.9.3 包封率和载药量的测定 按最优处方制备Jug/BSP-VES胶束溶液,测定其包封率和载药量,方法同“2.5”项。结果发现Jug/BSP-VES胶束溶液的包封率为(89.140±1.163)%(n=3),载药量为(6.493±0.087)%(n=3)。 2.9.4 粒径及ζ电位测定 按最优处方制备Jug/ BSP-VES胶束溶液,Zetasizer Nano ZSE纳米粒度电位仪测定其粒径、粒度分布及ζ电位。结果如图8所示,测得Jug/BSP-VES胶束溶液的平均粒径为(120.30±2.80)nm,PDI为0.169±0.014,ζ电位为(?27.00±1.25)mV。 图片 2.9.5 差示扫描量热法(differential scanning calorimetry,DSC) 分别称取适量胡桃醌、BSP- VES、胡桃醌原料药物理混合物和Jug/BSP-VES胶束样品置于铝制样品盘中压制,氮气为保护气,扫描范围25~350 ℃,加热速率10 ℃/min。结果如图9所示。胡桃醌的特征吸收峰在156 ℃,BSP-VES的特征吸收峰为184 ℃,与胡桃醌的特征峰不重叠;物理混合物中,二者特征峰均出现,而Jug/ BSP-VES胶束的热量曲线上无胡桃醌的特征峰,说明胡桃醌已被成功包载进载体,特征吸收峰消失。 图片 2.9.6 储存稳定性考察 按最优处方制备Jug/ BSP-VES胶束溶液,在pH 4.5,4 ℃和25 ℃条件下测定其在第1、3、7、15 d的粒径和包封率。结果如表9所示,在4 ℃下,Jug/BSP-VES的粒径和包封率无较大变化,说明储存稳定性较好;在25 ℃下储存效果相对较差,随时间增加,胶束溶液粒径变大,包封率降低,因此4 ℃为Jug/BSP-VES胶束溶液的最优储存条件。 图片 2.9.7 体外释放考察 采用透析法考察胡桃醌和Jug/BSP-VES胶束溶液的体外释药情况。分别将胡桃醌、Jug/BSP-VES胶束溶液置于透析袋(截留相对分子质量3 500)中,透析袋两端夹紧,分别浸没在含有0.5%聚山梨酯-80的醋酸-醋酸钠缓冲液(Ph 4.5)中;恒温水浴(37.0±0.5)℃,转速100 r/min,每组平行进行3组试验,分别于选定的时间点收集5 mL样品,收集后补加等量同温的释放介质,所得到的样品经微孔滤膜滤过后进行HPLC分析,体外释药曲线如图10所示。胡桃醌溶液在6 h时释放到80%左右,Jug/BSP-VES胶束在48 h时的释放率为(82.13±2.51)%,达到了明显的缓释作用,表明将原料药制备成胶束可减缓药物的释放速度。 图片 3 讨论 胡桃醌作为抗肿瘤活性成分具有一定的毒性,对金鱼的半数致死量(median lethal dose,LD50)为1.3 mg/L,对小鼠ig给药、ip的LD50值分别为2.5、25.0 mg/kg[19-20]。此外,胡桃醌及其代谢产物能与肾脏细胞溶质蛋白共价结合,造成肾脏毒性[21]。研究表明,酒精能使胡桃醌中的毒性成分转变为其他物质[22],以酒精作为溶剂的胡桃醌制剂通常不显毒性。本研究通过BSP与VES发生酯化反应成功制备了BSP-VES胶束,以胡桃醌为模型药物,通过溶剂挥发法制备了Jug/BSP-VES载药胶束。Jug/ BSP-VES载药胶束外观呈类球型,粒度测定结果显示,Jug/BSP-VES胶束溶液的粒径图显示峰形呈单峰,分布范围较窄,说明胶束溶液粒径均一。TEM下观察到的Jug/BSP-VES胶束,其粒径比粒度仪测定结果较小,可能是由于在制样过程中胶束水分的挥干导致粒子发生皱缩所致。BSP-VES作为两亲性高分子材料,在水相中的浓度超过临界胶束浓度后可形成胶束,制备方法简便。 本实验设计了一种可提高胡桃醌稳定性的载药胶束,拟制成温敏凝胶剂、采用阴道给药的方式,用于治疗阴道炎症、宫颈癌术后等。正常人体阴道pH值范围在3.5~4.8[23],因此,体外释放实验采用的是pH 4.5并含有0.5%聚山梨酯-80的醋酸-醋酸盐缓冲液[24],来模拟阴道中的酸性环境。在稳定性研究中,也重点考察了上述条件下载药胶束的储存稳定性,而并未采用通常的PBS(0.01 mol/L,pH 7.4)缓冲体系和含10% FBS的PBS(0.01 mol/L,pH 7.4)缓冲体系。另外,本实验所制备的Jug/BSP-VES载药胶束处方中尽可能减少了辅料种类,以避免腔道用药过程中的副作用及不良反应。 在单因素实验中,本实验考察各因素对处方工艺的影响。制备温度的高低主要影响有机溶剂除去的速度,温度过高或过低,引起有机溶剂挥发速度过快或过慢,均不利于胶束对药物的包载[25]。因考虑到温度对制备的影响较大,在25~35 ℃时Jug/ BSP-VES胶束中的胡桃醌含量不稳定,因此,对制备温度作进一步实验。 对有机溶剂用量的考察中,有机溶剂用量过少时,容易造成药物不能完全溶解,随着有机溶剂用量的增加,药物在溶剂中均匀分散,能与胶束较好地结合,当有机溶剂用量过多时,在有限的时间内,容易造成挥发不完全导致包封率降低[25],因此选择3 mL作为有机溶剂用量。 综上所述,本研究制备的Jug/BSP-VES胶束,通过单因素实验与CCD-RSM优化后,包封率好,粒径均一,稳定性良好,为胡桃醌制剂的应用开发奠定了基础。
如果胶束表面带有电荷,AFM可以探测胶束和基底的静电作用吗
请问那位知道用等温滴定微量热法测表面活性剂的胶束生成焓的方法
请问大家平时怎么表征胶粘剂的触变性,用旋转流变仪的话怎么表征,是做触变环吗
看到一篇单分子胶束的文献,里面说未溶剂化的链段(就是胶束中不溶的链段)不会有核磁信号,不太明白。 小弱觉得即使未溶剂化,测量时也处在同样的磁场中,应该也会有信号吧。 附件中是具体的说明。
今天请教一位博士师兄,他建议我用投射电镜表征,染色即可。对此不熟悉,特来请教。我做的是MMA-St-MAA-BA的种子乳液共聚制备核壳乳胶粒。谢谢大家了。
各位坛友好,最近本人在做有关反胶束的课题,发现能查阅到的制剂方面的文献很少,一般都是材料化学方面的,而在陆彬主编的《药物新技术与新剂型》中也是一语带过,很棘手,希望论坛中有研究过,做过这方面课题的战友能够提供一些帮助,能有文献参考,非常感激!
最近做CE分离中药中生物碱成分,用区带电泳分了很久,无果,于是想采用胶束电泳来分离。可是凡我缓冲液中加了SDs,不管多大浓度,都不峰,有事吼还会出现很难看的倒峰,即使出缝了,信号也小的接近于基线了,这是为什么啊?查文献中别人做的胶束电泳,峰都分得很好
乳液的浊度是否就一定随着胶束的聚集而增大吗?我配了一种溶液,它是由一种表面活性剂和AgNO3配制而成的,当AgNO3的浓度增大时,溶液变浑浊 。而且溶液中有BF4-离子,我最初认为是AgBF4在水中的溶解度不大。但是通过查资料说其溶于水。所以变浑浊是否应该认为是胶束的形成的缘故?
第一次做TEM,不是很懂,请大家帮忙分析一下下面的谱图,谢谢.我的样品是分子量5000的PEG-PLGA胶束,配成0.5mg/ml的水溶液,在碳膜铜网上大概滴5ul,快干时滴1%磷钨酸染色两分钟后,吸去多余的液滴,干燥后TEM观察问题1,磷钨酸负染法是不是亲水部分染成白色,疏水部分染成黑色,那背景就应该是白色的了,可是我看到好多别人做胶束的谱图背景都是黑色的,到底应该是怎样的颜色。问题2,谱图中的哪种结构才是我要的胶束,那些亮晶晶的小白点是不是磷钨酸?问题3,两张图都是在10W倍的放大率下拍摄的,位置也差不多,只是曝光时间不一样,怎么两张图差别那么大啊[img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001071023_194753_1920968_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001071024_194754_1920968_3.jpg[/img]
各位大神,求助一个问题:MEEKC的胶束标记物总不出峰怎么解决?多谢!
纳米材料的概述、制备及其结构表征是一个小型的综述[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=63065]纳米材料的概述、制备及其结构表征[/url]
我实验中采用的是硼酸-SDS分离介质,可是胶束的迁移时间怎么测啊?谢谢!
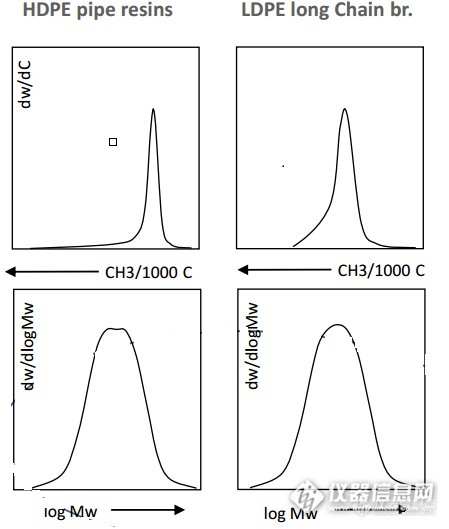
聚烯烃是一个复杂的体系,表征聚烯烃的方法有很多,比如用凝胶色谱分析仪表征分子量及其分布的信息,用化学组分分析仪表征其化学组分分布信息等,但是各种表征方法关注的点是不同的,分子量表征只关注聚烯烃链的长短或者说大小,而化学组分分布仪只关注聚烯烃支链的分布情况,支链越多,低温组分越多。这样也就会导致有时分子量相同的树脂,化学组分分布未必相同;有时化学组分分布相似,而分子量却差别很大,比如:http://ng1.17img.cn/bbsfiles/images/2016/01/201601180927_582163_1664_3.jpg上图中两种树脂管式的HDPE和长支链LDPE化学组分分布相似,而分子量分布确有不同,我们可以看到管式的HDPE分子量分布图封顶有一个平台。有没有能同时测得聚烯烃的分子量信息和化学组分分布信息的仪器呢?今天我就简单就给大家介绍下西班牙Polymer Char公司的聚烯烃多功能分析表征仪CFC:CFC是应用升温淋洗分级技术和凝胶渗透色谱技术的一台联用全自动分析仪,可现实双变量分布测定,首先按照结晶能力的不同,通过TREF分级,然后分级组分进入凝胶色谱柱,按分级组分的分子量进一步分离,进入到相应的红外检测器,根据测量结果可生成以温度和分子量对数为变量的三维谱图。在较短时间内完成复杂的共聚单体和分子量分布的充分表征,并且在分析过程中无需人工操作仪器。通过控制分析条件和样品加入量,CCD和MWD两方面的数据都可以达到很高的分辨率。CFC就像一个超级显微镜一样,能将聚烯烃的微观结构看的清清楚楚。
【序号】:2【作者】:祁凤英1,2王蕾2李东东2【题名】:可缓释富血小板血浆生长因子的新型自组装多肽水凝胶制备及性能表征【期刊】:中国组织工程研究 . 【年、卷、期、起止页码】:2024 ,28 (15) 【全文链接】:
我用的是硼酸-SDS,具体是100mM的硼酸,30mM的SDS,6%正丙醇,pH8.5,可是配好的缓冲溶液马上就会产生很多胶束,一进到电泳里电流马上就短流了,是我在配的时候有问题吗?还请高手指教[em0812]具体的配制是:50毫升的缓冲溶液,其中称了1.9克的硼酸钠,0.19克SDS,3毫升丙醇,用氢氧化钠调节的pH
最近写的一篇文章,主要是将激光共聚焦显微镜应用到磨损表征领域,因为其拥有数字化描述功能,相比扫描电镜具有一定优势。希望大家看后能提提意见,我准备找个杂志投一投,呵呵!见笑!
膜模拟化学 胶束、微乳、单层、双层、泡囊、主-客体系和聚离子的特性及应用[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=16010]膜模拟化学 胶束、微乳、单层、双层、泡囊、主-客体系和聚离子的特性及应用[/url]
[b]【序号】:1【作者】:[font=&][size=13px][color=#666666][/color][/size][/font][b][/b][url=https://search.cnki.com.cn/Search/Result?author=%E5%88%98%E5%BC%BA]刘强[/url][font=宋体][size=12px] [/size][/font][url=https://search.cnki.com.cn/Search/Result?author=%E9%83%AD%E6%AF%85]郭毅[/url][font=宋体][size=12px] [/size][/font][url=https://search.cnki.com.cn/Search/Result?author=%E5%BC%A0%E6%B5%A9%E6%98%8E]张浩明[/url][font=宋体][size=12px] [/size][/font][font=&][size=13px][color=#666666][/color][/size][/font]【题名】:[b][url=https://wenku.baidu.com/view/e5c296f0f8d6195f312b3169a45177232e60e470?fr=xueshu_top][b]气凝胶对聚氨酯硬质泡沫性能影响的研究与表征[/b][/url][/b]【期刊】:[font=&][size=13px][color=#666666][url=https://www.zhangqiaokeyan.com/journal-cn-13389/]《中国聚氨酯工业协会第十八次年会论文集》[/url][/color][/size][/font]【年、卷、期、起止页码】:[font=&][size=13px][color=#666666]|[b][url=https://cpfd.cnki.com.cn/Area/CPFDCONFArticleList-JAZG201607001.htm]2016年[/url][/b][/color][/size][/font]【全文链接】:[url=https://cpfd.cnki.com.cn/Article/CPFDTOTAL-JAZG201607001043.htm]气凝胶对聚氨酯硬质泡沫性能影响的研究与表征--《中国聚氨酯工业协会第十八次年会论文集》2016年 (cnki.com.cn)[/url][/b]
网上说布鲁斯特角显微镜大多用于空气水界面的单分子层的表征,想问一下,如果将铸膜液涂在玻璃基质上,可否用布鲁斯特角显微镜在线观察其内部微团的形态变化?
各位仪器行业前辈好!本人想进入科学仪器行业,希望有识之士引荐,小弟不胜感激!简单介绍个人情况:在我硕博阶段六年的材料研究过程中,XRD、AFM(NT-MDT Solver P47, Vecco D3100)是我常用到的表征技术:1. AFM方面,我熟悉AFM多通道检测技术,如:CAFM, MFM, SKM, LFM, PFM(组内都是我自己首次开发使用)。特别在PFM测试上,由于研究需要,我经常会使用到Asylum Research MFP3D, 并掌握各种分析技巧;2. XRD方面,我用到过不同型号的XRD仪器,比如荷兰帕纳科Panalytical X’pert Pro、日本理学Rigaku、英国Bede公司D1高分辨多功能衍射仪、以及基于国内三大同步辐射光源(上海光源,合肥同步辐射以及北京同步辐射装置)的XRD (德国Huber 5020六圆衍射仪)等。这些仪器我都是亲自操作,XRD原理自然也非常熟悉,精通各种测量模式:1.全谱2θ−θ和掠入射;2. omega摇摆;3.phi扫描;4.In-plane(2θ-Chi);5.倒空间Reciprocal Space Mapping等,此外我还做过变温XRD(高达925度),因此XRD测试经验比较丰富、分析能力自认为还较强。 我擅长的表征技术并非仅限于此,其它诸如Raman,UV-Vis,XPS等我也都比较熟悉。正是基于我博士阶段工作的经历以及我对物理仪器分析行业的浓厚兴趣,因此我希望能得到各位前辈的推荐,谢谢!zfu@semi.ac.cn
何为细胞外泌体? 外泌体最早发现于体外培养的绵羊红细胞上清液中,是细胞主动分泌的大小较为均一,直径为40~100纳米,密度1.10~1.18 g/ml的囊泡样小体。细胞外泌体携带多种蛋白质、mRNA、miRNA,参与细胞通讯、细胞迁移、血管新生和肿瘤细胞生长等过程并且有可能成为药物的天然载体,应用于临床治疗。 然而,测量技术手段的局限限制了外泌体在这些领域的研究进展。所以,在这篇文章中,作者总结了外泌体的纯化方法(离心法、过滤离心法、密度梯度离心法、免疫磁珠法以及色谱法),比较了现存各种外泌体测量技术(电子显微镜、动态光散射技术及纳米微粒追踪分析术)在外泌体尺寸和表征研究中的应用。原文点击——综述:细胞外泌体颗粒表征测量技术新进展
【序号】:1【作者】: 张亚南【题名】:聚电解质复合物纳米胶束的合成及性能研究【期刊】:江南大学 【年、卷、期、起止页码】:2015【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201501&filename=1014370567.nh&uniplatform=NZKPT&v=N8nmz_MTeIseDPCQglHFnPjs2A0-D2ZMflhoJYxzpAWDFqMpWg7IUqNHLX8MEBZH
[font=微软雅黑][font=微软雅黑]粉体流动性表征的是粉体在某些特定条件下的流动能力,药用粉体的流动性表征方法主要有[/font]3种:[/font][font=微软雅黑]1[/font][font=微软雅黑]基于测量颗粒质量流量的方法,包括霍尔流量计测量、低速转鼓中的颗粒物质的坍塌规模或质量流率测量等;[/font][font=微软雅黑]2[/font][font=微软雅黑][font=微软雅黑]基于测量颗粒摩擦的方法,包括静力学休止角、剪切流变力学、压缩度测量等[/font] [/font][font=微软雅黑] [/font][font=微软雅黑]3[/font][font=微软雅黑][font=微软雅黑]基于颗粒形状的分形维数法等[/font] [font=微软雅黑]。[/font][/font]
我想选择芘做荧光探针测量聚合物的临界胶束浓度,需要买一些,也不知道有没有国产的,希望大家能告之一二。
哪里有卖卖静电离子色谱柱,涂敷有胆汁酸诱导体胶束
请问,透析高聚物胶束应用何种型号的透析袋,高聚物分子量为3000-8000。谢谢!
【序号】:1【作者】:孙陆军【题名】:磷酸钙/明胶多孔支架的制备与表征【期刊】:【年、卷、期、起止页码】: 北京化工大学, 高分子化学及物理, 2008, 硕士【全文链接】:http://www.cnki.net/KCMS/detail/detail.aspx?QueryID=1&CurRec=43&recid=&filename=1014450337.nh&dbname=CMFD201501&dbcode=CMFD&pr=&urlid=&yx=&v=Mjc0NDZZUzdEaDFUM3FUcldNMUZyQ1VSTHlmWnVScUZpRG1WcnJJVkYyNkdyZTlIdExQcUpFYlBJUjhlWDFMdXg=
去年发表了一篇通过测试表面张力表征水玻璃老化的文献调研,试验起来发现效果并不理想。近期改用加速试验做了一些水玻璃抗老化剂的性能表征,特此与大家分享。试验原理:加速试验是指在保证不改变产品失效机理的前提下,通过强化试验条件,使受试产品加速失效,以便在较短时间内获得必要信息,来评估产品在正常条件下的可靠性或寿命指标.通过加速试验,可迅速查明产品的失效原因,快速评定产品的可靠性指标。(来自百度) 由于没有具体文献可以参考,所以为了找到最优的试验方案,做了大量的尝试,也有一些意外收获。试验方法:将添加了防老化剂的水玻璃稀释一定倍数,然后用硫酸中和至中性,观察不同配方发生凝胶的时间,从而判断防老化性的优劣(发生凝胶的时间越长,则产品越好)。首先寻找较合适的稀释倍数:先试了10倍,凝胶速度太快了,后面的酸还没加完,前面的已经凝胶了。再试100倍,太慢了,空白试验过了4小时都没凝胶,更别提加了防老化剂的了。30倍,还是太慢。只剩20倍了,试验发现20倍效果确实较好,空白的凝胶时间大约在半小时,有利于观察。中和这一步破费了些周折。我先是用酚酞做指示剂,加酸直到体系变成无色为终点。水玻璃中的硅酸根具有一定的缓冲能力,原本已经无色了,后来慢慢的又出来点红色。这也使我发现,pH的调节不够精确,因为最后各处理的颜色并不一致。这也导致了试验误差:本来有几个处理,防老化剂用量是递增的,用量最高的反而先凝胶,从指示剂颜色看是由于该处理pH偏低所致。因此,加酸方式得改!还是稀释20倍,改用移液枪迅速加入过量的酸(每个处理用的酸量都一样),使pH达到5的样子,小样,酸的量是一致的,而且都过量,你总没话说了吧!奇怪的事情出现了,所有的处理都不凝胶,包括空白在内,过夜还是流动的。百思不得其解!这个问题困扰了整整两天,做了五六批验证试验,终于发现,是自己想当然了!先想着酸过量总会凝胶的,而事实是,酸太多了也不行!只有在近中性时才会较快速的凝胶。基本搞定:较优化的试验方式:水玻璃稀释20倍,加等量的酸使体系pH值在7-8之间。
表征氮化硅微粉体个人有些地方有些迷惑,不知道选用什么方法和设备1、粒度方法:光散射法、沉降法、电导法?还有其他不清楚个人想选用 激光粒度仪,理由是检测快速,效率高,精度?2、比表面积这个不太了解,用什么方法和设备比较合适,xdjm帮忙推荐下3、元素分析主要分析金属元素Fe、Al、Ca等,氧、氮、游离硅含量、二氧化硅、氮化硅含量这个好像很复杂,选用什么方法测这些呢,我个人想法是ICP-AES(OES)测量金属元素,氧氮含量怎么测呢?红外氧氮分析仪?游离硅这么测呢,氮化硅含量怎么测量啊?4、堆积密度、堆积角?大家讨论讨论,陶瓷粉体表征有经验的xdjm给指导下







 我要推广仪器
我要推广仪器
 下载APP
下载APP