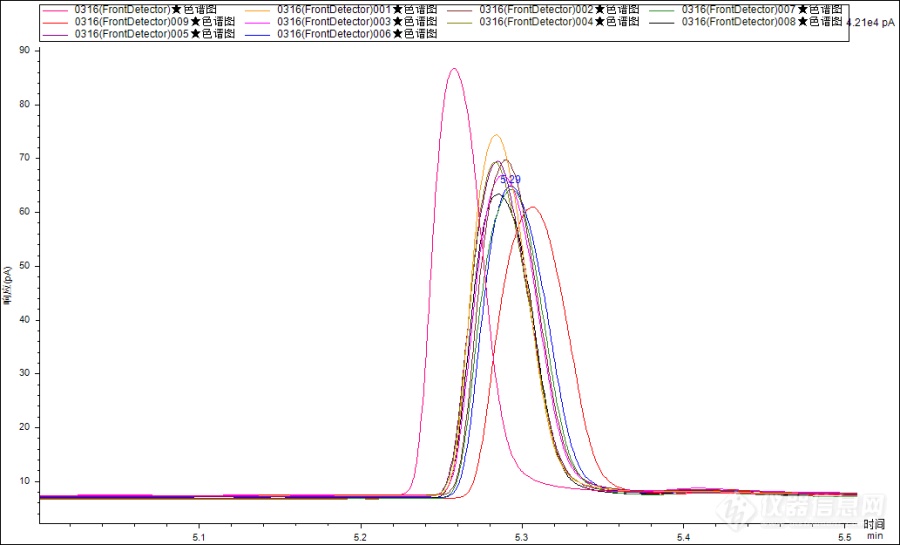
分析甲基苯丙胺时,连续进样时色谱峰峰型总是不理想,保留时间和面积重复性也很差,该如何解决。[img=,554,335]https://ng1.17img.cn/bbsfiles/images/2023/03/202303221438312389_8002_3913975_3.png!w690x417.jpg[/img]
使用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]检验甲基苯丙胺,突然峰出的很差,低浓度直接没有峰了,其他东西正常。
请教苯丙氨酸和苯丙胺醇的液相分析条件?有作过类似分析的吗?我用C18柱,未找到合适的方法,请有经验的同行不吝赐教。谢谢![em01]
甲苯 或甲基丙烯酸甲酯在水溶液中怎么用液相怎么分啊
安捷伦8860 5977b机子老化色谱柱以后,苯丙胺类标准物质不出峰了是什么情况啊,哪位大神可以帮助我一下

用亚甲基蓝测硫化物的时候,总发现空白偏高,0.06-0.08之间。多次排查觉得可能是对氨基二甲基苯胺盐酸盐的原因,下图是照片,品牌是adamas,外观为黑色,且大多结块。(我看网上都是说是白色至黄色至灰色,如果是黑色说明变质了问了售后,他们说这批对氨基二甲基苯胺盐酸盐就是黑色的,成品颜色跟工艺有关,还给了检测核磁和[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]的鉴定结果数据(但我看不懂,放在附件了),说这批药的结构是合格的。我现在的疑问是,即便这批药的结构是合格的,但是不同生产工艺造成的成品颜色是否会影响空白样的显色呢?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/10/202110261016565152_2614_5383916_3.jpg!w690x920.jpg[/img]这个是对氨基二甲基苯胺盐酸盐的外观[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/10/202110261017246679_49_5383916_3.jpg!w690x920.jpg[/img]这个是配置好的对氨基二甲基苯胺溶液的颜色(是不是有点 深?)
请问老师,如果用光谱的方法进行盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量的测定,它们的吸收峰是多少???
请问老师:如何分析盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量??含量在几个到几十个PPM
测定Al用的六次甲基四胺溶液,40%,应该称40g溶解后定溶于100ml?还是直接溶解于100ml水中?需不需要加入盐酸呢?
请问各位大虾:结晶紫指示液、萘酚苯甲醇指示液、甲基橙的饱和丙酮溶液、高氯酸滴定液的保存期限各是多少?谢谢了
测定多环芳烃,用六甲基苯溶液作内标,购买的六甲基苯是固体的,请教溶液怎样配?是不是用二氯甲烷做溶剂?具体怎样操作,溶质和溶剂的量是多少?E-mail:zhoujuan@stu.xjtu.edu.cn
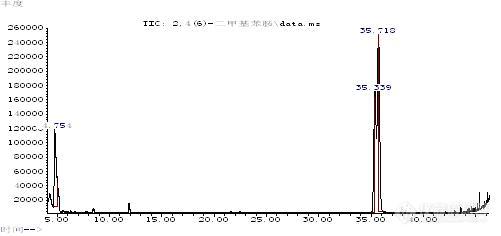
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
各位用3355四甲基联苯胺测余氯的同行:有几个问题欢迎大家讨论:1.你们用的3355四甲基联苯胺溶液是按GB5750.11-2006标准配置的吗?配制时有将其中的0.1mol/lHCL浓度加大的没有?2.你们的水样PH一般是多少?有需要调整的吗?大概PH多少时需要调整?3.调整时是先加1+4HCL再加水样调节好PH后再加入3355四甲基联苯胺溶液,还是将1+4HCL和3355四甲基联苯胺两种一起加入比色管再加入水样比色的?或是直接使用已加大HCL浓度配制的3355四甲基联苯胺溶液再加入水样检测?4.有做过用3355四甲基联苯胺比色法和HACH便携式或在线监测(DPD试剂)检测法对照试验的吗?两种方法测定结果怎样?谢谢各位发表高见!
求问,N,N二甲基对苯二胺盐酸盐,我这里是紫黑色,配制完大概是深灰色,我现在做空白高,不知道是不是这个试剂的问题?我看网上有说这试剂应为白色或灰色,灰色的话可是浓度高了?
求,分析有机溶液(PTA对苯二甲酸)中对甲基苯甲酸(PT酸)和对羧基苯甲醛(4-CBA)含量的方法,液相也行,最好为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法,当然有滴定的方法,更加,但需测试微量,就是只有150PPM以内的,谢谢了
要用分光光度计测二甲基甲酰胺溶液的浓度,请问标准溶液如何配制,怎么标定呢?
领导让做聚丙烯酰胺的离子度,标准17514-2017里面提到甲基乙二醇甲壳素(MGC)标准溶液0.005mol/L,请问我要怎么采购,供应商那里说没有这东西,只有甲基乙二醇壳聚糖
配制完N,N二甲基对苯二胺,溶液应该呈紫色,为什么这次溶液呈透明淡黄色?
我们化验室用3.3'.5.5'-四甲基联苯胺做余氯,(GB5749-2006的余氯检测方法)请问各位,我们配制3.3'.5.5'-四甲基联苯胺溶液时,溶液很快就变成黄色。这样可以使用吗?还有就是,当用此溶液检测余氯时,显示为黄绿色,为什么会这样呢?
请问甲基纤维素溶液怎么才能配制成透明的?我配的3%的,先把60ml水加热到60度,边加甲基纤维素边搅拌,然后加入4度的水40ml,放在冰上搅拌,溶液不是透明的,而且有很多气泡。
各位大神,大家好。我要测试水溶液中的间苯二胺的浓度,这个气相色谱之前没有接触过。问之前的做过的,他直接水稀释进样。但貌似水溶液直接进样对仪器有损伤。请教大神,这个需要设计一个怎样的实验方案。我想萃取的话,萃取出来的间苯二胺就可以用乙醇作溶剂,若果萃取的话,要选择合适的萃取剂。主要是在线监测使用。谢谢大家。
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]基线不稳,测得是丙酮-苯酐溶液
丙烯酰胺29g,N,N'-亚甲基双丙烯酰胺1g,定溶至100ml,室温和37℃搅拌都不溶解,怎么回事啊?可能原因是什么?是药品变质了吗?
我买的是2ug/ml的苯中甲基汞溶液,用原子荧光做形态分析,请问大家用什么溶剂稀释,做工作溶液,甲基汞的溶解性质如何?
各位专家,谁能赐教分析下列溶液组分中的苯胺和DAM含量可以采用什么分析方法?溶液组分:氯化钠、氢氧化钠、苯胺、DAM、环己胺、环己醇
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
测余氯时所用的是邻联甲苯胺溶液,但此试剂很难溶于水,搅拌的效果也不好,请问还有什么方法能快速溶解邻联甲苯胺?
国民“妖精”妲己也逃不过朝阳区群众雪亮的眼睛啊,竟也被举报了,于是,有人调侃说,“啊,监狱风云女主角已到位,可以开拍了!”明星为何都爱吸毒?所谓的毒到底是什么?实验猿告诉你!科普时间到!冰毒又名甲基安非他明、去氧麻黄碱,是一种无味或微有苦味的透明结晶体,纯品很像冰糖,形似冰,故俗称冰毒。吸、贩毒者也称之为“冰”。有胶囊、粉剂、小块等多种形式,可抽吸、鼻吸、口服或注射。该药小剂量时有短暂的兴奋抗疲劳作用,故其丸剂又有"大力丸"之称。http://ng1.17img.cn/bbsfiles/images/2016/03/201603071620_586178_2452211_3.png需要注意的是,该物质主体化学成分为甲基苯丙胺,但市面上的固体冰毒实际上是甲基苯丙胺盐酸盐,因此冰毒与甲基苯丙胺并不能完全等同。广义上的冰毒制品还包括摇头丸。为何明星爱“冰”?很多明星用来保持身材的法宝啊,喂!1887年,罗马尼亚化学家 Edeleano 在德国柏林首次合成了麻黄素的类似物苯丙胺。同年,日本化学家长井长义也从麻黄素中结晶提取出了苯丙胺,最早曾用于治疗肥胖症,因其可以保持头脑清醒并抑制食欲(至今治疗肥胖症的药物中也有苯胺类药物)。吃完就能上天堂啊!苯胺类药物的兴奋作用很早就被认知。由于苯丙胺具有较强的短时间内提升脑力作用,在第二次世界大战时,多在士兵军队内推广使用,还有报道称日本的“神风特攻队”的悍不畏死部分原因是由于使用了兴奋剂。随后大量苯丙胺从军队流入民间,日本战败后,苯丙胺类药物成了日本当时最好的心理药物。这可难不倒实验猿们?20世纪70年代中期,日本的毒贩将盐酸取代硫酸进行固体加工,无意中制造出不再是以往的粉剂状,而是如冰块一样晶莹剔透的固体晶状物。这种固体甲基苯丙胺,即甲基苯丙胺盐酸盐。也就是当下通用的冰毒。http://ng1.17img.cn/bbsfiles/images/2016/03/201603071621_586179_2452211_3.png需要强调,冰毒的主体成分或者说发挥作用的是苯丙胺类物质。制备方法:麻黄素HI/红磷还原法(又称Emde法)曾是被制毒分子广泛采用的冰毒合成方法。将麻黄碱在催化剂作用下加氢还原就可以变成冰毒。早期报道的冰毒主要集中于这种方法。用这种方法据说可获得纯度接近100%的冰毒。如何选择检测方法?检测方法主要采用气相色谱法(GC)、液相色谱法(HPLC)、毛细管电泳法、气相色谱-质谱联用法(GC-MS)、液相色谱-质谱联用法(HPLC-MS)、放射免疫法、胶体金层析法、免疫分析法等。传统的前处理主要是液-液萃取和固相萃取方法。近些年,固相微萃取、液相微萃取和微波萃取技术已经广泛应用于法庭科学领域。http://ng1.17img.cn/bbsfiles/images/2016/03/201603071622_586180_2452211_3.png但检测中有一个难点就是试样的分离提纯,冰毒制品经常混有麻黄碱或类麻黄碱,而麻黄碱主要包括麻黄碱、伪麻黄碱、甲基麻黄碱、甲基伪麻黄碱、去甲麻黄碱和去甲伪麻黄碱6种,它们在麻黄植物中都有一定的含量,而且互为三对同分异构体,认为最适合毒品复杂体系分离的毛细管胶束电动色谱(MEKC)也难以进行有效分离。1.薄层色谱法2.气相色谱法该法适用于带挥发性成分( 如苯丙胺)的检测,但对色谱条件有要求。由于固定相的吸附导致色谱峰常有脱尾和不对称现象,可以用氢氧化钠或氢氧化钾预处理色谱柱以增加固定液的极性来阻止胺的离子化。这两种色谱柱可以串联使用。检测时多将其进行衍生化处理,固定液多用聚甲基硅烷(SE-30)或者苯基甲基聚硅氧烷(OV-17)。3.色谱-质谱联用法该法(CG-MS)需要将检测对象转变成衍生物形式,如乙酰基或异硫氰酸盐衍生物,检测样本多为毛发。50mg毛发样对为0.05ng·mL-1。如丙基氯甲酸酯衍生化,用氘化的苯丙胺衍生物或N-丙基苯丙胺可以检测尿液中的冰毒代谢成分。4.高效液相色谱法该法(HPLC)使用的相对较少,由于冰毒的紫外吸收特性不好,但是该法可不对样品进行衍生化处理。检测之前需用β-葡糖苷酸酶和硫酸酯酶在37℃条件下水解24h,固相萃取后可用紫外检测器在215nm下测定。5.马改氏试剂检验6.拉曼谱图7.荧光光谱警察蜀黍怎么确定某人“溜冰”了没呢?检验一个人是否吸食病毒类毒品,通常是通过毛和体液。过去研究较多的是血、尿等体液类,但这两类检材只能反映收集样品前的1~3天中的毒品服用情况,而毛发可提供吸毒者的毒品使用史及使用程度的信息。但有个问题,它里面所含的毒品及其代谢物的含量一般很低(ng/mg级),而尿液或血液要高得多(lg/mL级),另外,毛发中的生物杂质含量很高,因此采用气相色谱分析时需要采用特殊的方法,如采用电子捕获负离子化学源(NICI),据称最小检测限可达413~ 9118pg/mg。另一常用的提高灵敏度的方法就是对样品进行衍生化处理,它对于改善色谱行为、提高检测灵敏度、获得特征离子峰、增强检测的灵敏度同样具有明显的作用。检测毛发中冰毒类毒品的常用方法是把毛发消解,然后经过固相、液相或超临界流体等技术对消解液进行萃取,用氮气将萃取液吹干后,在水浴加热条件下进行衍生化处理,然后采用GC等进行检测。来源:实验与分析
我们领导让测,聚丙烯酰胺,标准17514-2017里面提到的甲基乙二醇甲壳素标准溶液怎么配制?供应商说没有只有甲基乙二醇壳聚糖,这两个是一回事吗?请教各位大神老师们,万分感谢[img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905171326298076_8329_3493743_3.png[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP