我是搞地质化验的,有化验痕量金这一项,可是我不知道化验痕量金的目的。我们化验室做痕量金用光谱做半定量分析,大家都是用什么方法啊?
我想看看样品中一种痕量元素和Fe,Al的分布关系,打算用SEM或者TEM,但突然想到痕量元素本身含量很低,不知道是否能否检测到!请问大家mapping扫描痕量元素含量至少需要多少才能有效?那XRF-Map呢?
岛津GC-2014C的仪器上,在SPL进样口的进样模式里面有不分流进样模式,不分流进样模式是针对于痕量化合物组分的分析,这个痕量有没有标准呢?多少个ppm才算是痕量呢?
来化验室两年,由于是地质单位化验室,所以总是在做痕量金,可是根本不知道做它的目的何在,有什么用?
来化验室两年,由于是地质单位化验室,所以总是在做痕量金,可是根本不知道做它的目的何在,有什么用?
想请教各位一下石墨炉原子吸收测痕量金速度怎么样,一分钟测几个样啊?那个厂家的速度比较快,检出限比较低,望知道的帮帮忙哈,谢谢!
要做文章了,就不是不知道从哪方面入手,我是做石墨炉测定痕量金,不知道大家有什么好的思路!如果有几篇新的论文参考就更好了。
测定地质样品中的痕量金.不知道有没有新的方法,最新的?
在痕量,超痕量ICP分析中(纯度99.999%)对于环境,水质,溶剂(酸)有没有特殊的要求? 谁有这样的文献和相关资料, 麻烦发一份给我,谢谢!我的邮箱:gongmm@lsam.cn
ICP-MS做地质样品的痕量金,王水介质,能测定准确吗。有相关的操作规程最好。
目前采用石墨炉测金比较普遍,但是ICP-MS测定地质样品的痕量金,存在哪些缺陷或者短板,请大家讨论。
http://image.instrument.com.cn/bbs/UserForumdLive.asp?username=liuqi9999&threadid=3650742 来化验室两年,由于是地质单位化验室,所以总是在做痕量金,可是根本不知道做它的目的何在,有什么用?
摘 要:随着分析科学的不断发展,常用的元素分析方法,如光谱技术AES ,AFS) 和质谱等已不能满足环境和生物样品中痕量、超痕量元素的赋存形态分析。以色谱联用技术为代表的元素形态分析测试技术(如:液相色谱- 原子光谱联用、色谱- 电感耦合等离子质谱联用、毛细管电泳- 电喷雾离子化质谱联用技术等) 已成为国内外研究的热点。本文扼要的介绍了近年来国内外在环境和生物样品中痕量、超痕量元素砷、硒、汞形态分析的色谱联用技术研究进展,并侧重于样品前处理方法、痕量或超痕量元素的形态分析技术。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31681]色谱联用技术在环境和生物样品中痕量超痕量元素形态分析研究进展[/url]
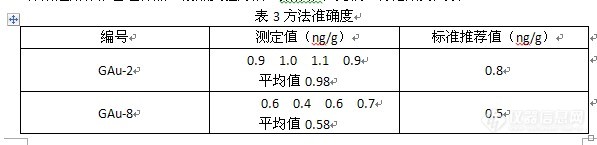
痕量金标准物质湿法分析研究摘要:本文采用王水封闭溶样,活性碳吸附柱原理,将泡沫塑料挤入玻璃吸附柱颈内,将痕量金标准样品溶液进行动态过滤吸附,吸附后的泡沫塑料直接用硫脲解脱,解脱后的溶液用石墨测定。方法精密度再7.1%,准确度满足地质实验室痕量金标准物质质量要求。关键词:痕量金标准物质,动态吸附,泡沫塑料金的泡沫塑料吸附富集分离已是金的富集分离法中应用最广泛的方法之一。泡沫塑料吸附石墨测定痕量金是分析痕量金中最常用的方法,该方法操作简单,效率高,已广泛应用于地质实验实验室中。在常规的预处理过程中,泡沫塑料直接放于试样溶液中振荡吸附,吸附率在80%左右,对于普通样品的测定,可能不会造成分析数据的超差,但是对于标准物质的测定,就很可能超差,鉴于此,本文在实验的基础上,采用活性炭吸附的原理,用泡沫塑料挤入吸附柱颈内动态吸附,实验证明,该方法具有更高的精密度和准确度。1 实验1.1主要仪器及试剂Z-5000型原子吸收仪动态吸附装置封闭溶样装置(水浴锅、聚四氟乙烯溶样罐,振荡器)盐酸、硝酸,氟化氢氨、抗坏血酸、硫脲均为分析纯金标准溶液(0.1µg/ml,10%王水介质)1.2实验方法称取10.0g样品,于高温炉650℃烧2个小时,将样品到倒入聚四氟乙烯溶样罐中,加入少量氟化氢氨润湿,加入25ml王水,盖上盖,振荡摇匀,放入水浴锅中水浴90分钟。冷至室温,加入70-80ml水。倒入预先装好的吸附柱中动态吸附,吸附后的泡沫塑料,放入预先加有1%的硫脲的试管中,水浴40分钟,挤出泡沫塑料,溶液上石墨炉测定。1.3标准系列配制移取0,0.2、0.5,1,2ml金标准溶液(0.1µg/ml)于10ml试管中,用1%硫脲定容到10ml,这样得到0,2,5,10,20ng/ml的标准系列。配制好的标准,随样品放入水浴锅内水浴(空白除外)。1.4吸附柱的安装 将市售的泡沫塑料切成3×1×1cm,用10%盐酸煮沸10min,洗净,然后放入玻璃吸附柱的颈内,装上布氏漏斗,在漏斗上面放一张滤纸和一层滤纸浆。见图1。 图1 吸附装置http://ng1.17img.cn/bbsfiles/images/2011/07/201107032232_302914_1601823_3.jpg1.5仪器工作条件http://ng1.17img.cn/bbsfiles/images/2011/07/201107032239_302918_1601823_3.jpg2结果与讨论2.1标准系列 配置一份标准系列,随样品一起水浴,采用吸光度法测定,制作曲线,测定结果见图2 图2 标准曲线http://ng1.17img.cn/bbsfiles/images/2011/07/201107032241_302921_1601823_3.jpg2.2普通吸附与动态吸附比较称取标准管理样,分别按照常规直接将泡沫塑料放入样品溶液中振荡吸附,动态吸附后,用石墨炉测定,将结果列入表1中。http://ng1.17img.cn/bbsfiles/images/2011/07/201107032242_302922_1601823_3.jpg从表1可以看出,采用动态吸附柱,由于吸附过程加入了过滤装置,泡沫塑料吸附的是没有残渣的溶液,吸附率明显提高,且与标准推荐值很接近。2.3方法精密度称取标准管理样品GAu-9,12份,按照实验方法,用石墨炉原子吸收测定,计算标准偏差,结果列入表2中。http://ng1.17img.cn/bbsfiles/images/2011/07/201107032246_302924_1601823_3.jpg2.4方法准确度http://ng1.17img.cn/bbsfiles/images/2011/07/201107032247_302926_1601823_3.jpg3结论 采用常规的分析方法富集分离痕量金标准物质,常常出现测定值偏低的情况,采用封闭溶样,泡沫塑料动态吸附柱吸附,石墨炉测定痕量金标准物质,不仅提高了吸附率,而且大大提高了标准物质分析的准确度和精密度。
ICP-MS测定痕量金,处理方法,10g样品,王水溶解,泡沫塑料吸附,低温灰化,然后650度灰化,然后用王水蒸直近干,10%王水提取定容到10ML,移取1ML,用2%硝酸定容至25ml比色管中,方法是否可行,干扰如何消除?从质谱干扰和非质谱干扰两方面进行探讨。
[b][color=#cc0000]直读光谱分析常说“痕量分析”,说明材料里含的元素非常少,属微量级的,那么“微量”与“痕量”有多大区别呢?[/color][/b]
根据IFRA的信息通讯(IL 948),考虑科技、分析方法和监管的发展,IFRA认为对其关于日用香料中无法避免的痕量杂质问题的立场,需要进行重新定位。不同的法规体系,对“痕量水平”的定义不尽相同,而且分析方法的灵敏度不断提高,物质的检出限越来越低;但是,监管框架并未发生变化,生产者应确保痕量杂质的存在不影响原料和产品的使用安全。 考虑到安全,IFRA已经对一些日用原料中含量极低的杂质成分进行了限量,这是针对某些物质的具体情况而制定的,而不是关于痕量污染物的通用规定。据信,一些国家和地区的监管也采取这种方式。 IFRA认为对众多日用香料中的所有痕量物质制定一个单一的通用限量的观点并不恰当,也无必要。只有IFRA认为确有必要对某些物质(例如,苯、甲苯、黄樟素等)进行限制时,才会制定相应的IFRA标准。 任何对痕量物质的管理研究都应建立在各成员企业依据法规要求对单个物质的安全考量和预期使用的基础上。
有机进样系统进样用到的试剂,可是找不到生产痕量金属级的厂家,求推荐
目前手里有一支盲样是阿特拉津 333903,大家帮我看看是不是过期标样?还有值是多少?
做阿特拉津加标回收率时,萃取后用氮吹浓缩,温度和吹扫压力怎么设置才能尽量提高回收率呢
水中阿特拉津扩项时,旋蒸后阿特拉津回收率很低,求做过的指导一下怎么能降低损失,或者有没有降低损失的其他浓缩方法
水中阿特拉津扩项时,旋蒸后阿特拉津回收率很低,求做过的指导一下怎么能降低损失,或者有没有降低损失的其他浓缩方法
水中阿特拉津扩项时,旋蒸后阿特拉津回收率很低,求做过的指导一下怎么能降低损失,或者有没有降低损失的其他浓缩方法
各位亲我现在要用液相来测定土中阿特拉津的含量,需要用内标法,但是我是第一次接触到液相,什么都不懂请各位给我支一下招,看看有什么办法啊
大家MS方法设置[font=Arial, sans-serif][size=13px]痕量离子检测勾不[/size][/font]
如果用ICP测Te基体中的痕量Si元素应该怎样做(如样品前处理、仪器优化等等)?请各位帮忙想办法!
痕量元素是什么意思
常量、微量、痕量是如何规定的?在哪些文里有规定,好了快10年化验了都不知道,惭愧呀,望老师不吝赐教
石墨炉做痕量银,大家都用什么方法,麻烦给推荐一下,还有加不加基改。
各位好,最近我们用万[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]做痕量硫酸根,不知为什么,一旦浓度低于50 ppb,就会在硫酸根位置出负峰。后来咨询了仪器公司的工程师,他们开始说是我们实验室的水不好,后来借了其它实验室的饿Millipore的水,还是不行,后来他们又告诉我们不要用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]做痕量硫酸根,而应该用电位滴定,可是当时购买的时候明明说可以做到0.5 ppb的啊。我们用的是MIC,据说这是最好的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]了,为什么连硫酸盐都做不了啊?难道是我们实验室的水的问题吗?还是我们的水平问题?我们确实没有做过[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],不过以前做过液相的,应该差不多吧?做痕量硫酸盐需要注意什么呢?求各位大侠指点迷津!







 我要推广仪器
我要推广仪器
 下载APP
下载APP