胶体磨是流体或半流体物料通过高速相对连动的定齿与动齿之间,使物料受到强大的剪切力,磨擦力及高频振动等作用,有效地被粉碎、乳化、均质、混合,从而获得满意的精细加工的产品。1、食品工业:芦荟、菠萝、果茶、冰淇淋、月饼馅、奶油、果酱、果汁、大豆、豆酱、豆沙、花生奶、蛋白奶、豆奶、香精、各种饮料、芝麻、猪皮及其他动物皮等。2、医药工业:各型糖浆、营养液、中成药、膏状药剂、生物制品、鱼肝油、花粉、蜂皇浆、各种药膏、各种口服液等。3、日用化工:牙膏、洗涤剂、洗发精、鞋油、高级化妆品、沐浴精、肥皂、香脂等4、化学工业:油漆、颜料、染料、涂料、润滑油、润滑脂、柴油等。5、其它工业:塑料工业、纺织工业、造纸工业、生物化工、环保节能、纳米材料、各大中院校、科研单位等。
各位大侠,我用紫外检测器做烟嘧磺隆和吡嘧磺隆农药样品, 标样都没有问题,出峰很好。但是将农药制剂稀释后进样,完全没有峰了。也将标样直接添到制剂中再稀释,但是仍然没有峰出来。用液质质进样,该样品吡嘧磺隆是没有问题的,烟嘧磺隆也能出峰但有一定干扰,请教一下,液相紫外检测器做不出该农药样品这到底是什么问题呢?

我们用的是waters的ACQUITY UPLC/XevoR TQD,原来血浆样品处理是血浆100ul+内标液100ul+标品溶液100ul+100ul甲醇涡旋离心沉蛋白,然后直接取上清进样,流动相是甲醇:水,以前血浆样品都很正常,这周做的时候内标峰偏移厉害,如图上面是出问题的内标峰,下面是用水甲醇配的内标标品进样。求指导!http://ng1.17img.cn/bbsfiles/images/2014/12/201412041845_525919_2809725_3.jpg
我的仪器是安捷伦7890/5975,现在遇到个问题,大家帮忙解决下啊!我9月10号更换了新的侧板后调谐通过了(更换侧板之前已经清洗过离子源)推斥极22.31,当时工程师检测也说一切都正常,但是随着时间推移, 做样的丰度一天比一天降低,现在只有1000左右,调谐也可以通过,但是推斥极就到了34.81,EM电压也比之前高了200多,打给400让我清洗离子源,我照做了,还老化了柱子,清洗了进样口,更换了进样针、衬管、进样口隔垫、前级泵油,开机调谐还是通过了,但是推斥极还是一样,EM电压1059,降了200,可是做样品丰度还是很低,我把EM电压增加了200,再做样,样品的丰度3500-4500,还是很低啊!各位高手帮帮忙吧
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]小白求助,血浆样品中内标物质大黄素峰一直前移,但是待测物质峰保留时间不变,大黄素是用dmso溶解配成1mg/ml的母液,然后甲醇稀释成100ng/ml来使用的。血浆样品进样体积是10微升。第一张图是暑假之前做方法学验证的时候,那时候内标峰保留时间和峰面积一直很稳定。暑假时仪器请了工程师来维护,放假回来之后做内标峰就变丑了,如第二张图所示,但是峰面积和保留时间没有太大变化。最近做的时候,就出现第三张图这种,内标峰一直前移,而且峰面积也在变小。在这期间,色谱柱,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法,仪器,流动相都没有变动。尝试过每针加长平衡时间,但是没有太大改善,改变了一下[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]梯度,也没有改善,也换过另一个相同的旧柱子,实在没有头绪怎么解决了。请问各位大神这是怎么回事呀,是仪器问题还是色谱柱出问题了,或者是其他别的问题[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211202204185742_4923_5557266_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211202207338199_913_5557266_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211202207354951_2539_5557266_3.png[/img]
液相检验样品杂质,每个样品在一分钟处都出现个未知杂质,同一次运行序列的样品这个峰的峰面积都差不多,另外标准溶液中也有这个峰,峰面积和同次运行的样品相差不大,空白溶液中没有这个峰。想请教各位大神,这个杂质能否判定为降解峰?计算杂质总和时需要计入此峰吗?这个杂质的判定需要通过什么实验进行验证,以符合GMP的要求,审计官不会质疑?求解答!感谢!
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]跑样,血浆里有两个药物,同种样品处理方法,一起分析的,一个物质连续跑标曲质控,峰面积稳定,但另一个物质跑着跑着峰面积就下降了,跟标曲拟合不上,请问这是为什么?
液相检测样品杂质,每个样品在1分钟处都会出现一个未知杂质,并且同一次运行的样品这个杂质峰面积都差不多,标准溶液中也出现这个峰,峰面积和同次运行的样品也相差不大,空白溶液中没有该峰。想请教各位大神,这个杂质是否能够判定为降解峰?计算杂质总和是否需要计入此峰?对此峰的判定需要做什么实验以符合GMP的要求,审计官不会质疑,求解答!感谢!
[color=#444444]仪器:[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]AB5500[/color][color=#444444]流动相:乙腈-甲酸水[/color][color=#444444]梯度洗脱,出峰时间在88%等度洗脱中[/color][color=#444444]对于待测成分进行了回收率测试80%, 空白血浆加标准品走标曲r=0.996 ,[/color][color=#444444]其中两个成分出峰时间分别为3.0和5.70,无杂峰[/color][color=#444444]采集的血浆样品进样,一个成分在5.7min不再出峰,但于4.6和5.1min出了两个响应的峰[/color][color=#444444] 另一个成分在2.8min和3.0出峰,两个峰连在一起无法定量[/color][color=#444444] (上传的图自上而下依次是:空白血浆+标准品;采集样品;采集样品+标准品)[/color][color=#444444]现在这两个成分都无法定量,我觉得可能是其他成分代谢产物在同一通道出峰,影响了待测化合物的出峰,但是又分不开[/color][color=#444444]想求助大家,该怎么定量这两个化合物?[/color][color=#444444][img=,230,540]https://ng1.17img.cn/bbsfiles/images/2019/06/201906111123585259_1609_1843534_3.jpg!w230x540.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906111123589171_8887_1843534_3.jpg!w690x517.jpg[/img][/color]

样品是测定某溶液中牛黄胆酸钠含量,流动相为乙腈:04%磷酸(30:70),波长为203mn,仪器为Waters9针样品出峰时间和峰面积分别是出峰时间 峰面积11.668 25853211.690 26846611.732 26852911.760 26187211.779 27389911.801 23583611.840 23248911.866 23546611.901 233216 主峰不断地退后,但其他杂质峰位置不变,而且峰面积很规律下降?这种情况有什么办法解决?http://ng1.17img.cn/bbsfiles/images/2012/11/201211101435_402866_1919313_3.jpg
我在检测枸橼酸离子的时候标准品的溶剂峰是倒峰(溶剂峰为水)而样品测试时溶剂峰(磺基水杨酸)比样品峰要高出8倍左右,怎样处理才可以消除溶剂峰?检测条件是:流动相为硫酸,检测器是示差检测器.谢谢各位了.
血浆生物样品的峰分离度一般要求好多喃?最近峰分离度不是很好的
[align=center][size=24px]HS[/size][size=24px]-10[/size][size=24px]报“样品瓶升降器马达错误”的维修[/size][/align]故障现象:HS-10运行中报“样品瓶升降器马达错误”。当瓶子恒温完成,进入加压阶段时报错。[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309251909302420_9954_2592430_3.jpeg[/img]故障处理:HS-10的样品瓶升降器是在样品瓶加热完成后,将目标样品瓶向上顶,从而使进样针扎入瓶子中进行加压、导入等一系列动作;而在进样结束后,升降器下降,针座上的弹簧又将瓶子顶回。[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309251909308597_166_2592430_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309251909313675_6392_2592430_3.jpeg[/img]现场重现了故障,在上升过程将近完成之时,会有一声异响,怀疑是顶瓶的阻力过大造成。测试将底座和弹簧去掉时,样品瓶升降器上升的动作正常无报错无异响。观察压过的瓶子如下图所示,有弧形的压痕。而正常的情况下,瓶盖不会出现这样的偏向一边的压痕。[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309251909315835_779_2592430_3.jpeg[/img]用户描述曾经自己拆过进样针。但是查看进样针底座的三个螺丝只剩两个了,由此怀疑用户可能自行拆过底座,导致弹簧位置不正确,从而压瓶也出现了不正常的痕迹。将底座弹簧拆下,发现已不正。[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309251909317663_714_2592430_3.jpeg[/img]当弹簧位置正确时,瓶子上升过程中,弹簧会被压缩在底座中间部分的缝隙里,不会有太大的阻力。而当前弹簧位置歪斜了,瓶子上升时,弹簧被压缩,不能折叠在缝隙中,导致阻力过大从而马达报错。后来在更换了新的底座和弹簧组件之后,动作正常,也没有了异响。查看瓶子的压痕也正常。总结: 如果不确认拆卸或者维护的步骤是正确的,最好不要贸然行动,否则可能因小而失大。该用户本来是因为报“进样针未拔出”错误,打算拆进样针进行清理的,但是由于没有认真做准备工作,误拆了底座,又没有正确安装,致使弹簧错位,造成了更大的麻烦。
我做体内药物分析,血浆样品前处理步骤为血浆加入1M盐酸,然后加入乙酸乙酯,低速离心,再取上清吹干,再加入甲醇溶解,可是不知道为什么,溶解出的东西特别黄,与之前类似处理的结果不一样,感觉样品很杂很脏,哪位高人能告诉我是为什么啊?
降低样品的气化温度利于分子离子峰出现?是这样吗?气化温度的降低可以减少分子离子进一步断裂的可能性,分子离子峰的相对丰度增加。 如三十烷烃在 340℃时气化,不出现分子离子峰,改变 70℃气化时分子离子峰的丰度接近基 峰。
公司测试样品时,在高倍率的情况下会发现样品晃动。样品固定没有问题,象散也调好了。
HPLC测定柠檬黄时,标准液的出峰时间与样品加标后的出峰时间不吻合,为什么??我们的流动相为甲醇:乙酸胺=50:50,柱子为反相C18,流量为1 .5.
我的样品出峰时间是8.9,内标出峰5.4 反相色谱柱C18(150*4.6 ,5)梯度条件0-5min,10%乙腈。5-8min,10%-45%乙腈。8-15min,45%乙腈。流速是0.9现在我的主要问题是跑样品时,样品浓度越大,8-12min那段时间的基线越高,峰也很多很杂,但我的样品是8.9出峰。附图样品浓度0.3和300以及1000μg/mL在最大吸收波长下的色谱图[img=,690,794]https://ng1.17img.cn/bbsfiles/images/2021/05/202105240936143034_7645_5230299_3.png[/img][img=,690,856]https://ng1.17img.cn/bbsfiles/images/2021/05/202105240936148249_2126_5230299_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/05/202105240936152629_1823_5230299_3.png[/img]

作者:阿迪列提1,谢淑英2,陈勇3,李文霞4,向智敏3(1.阿勒泰地区药品检验所,新疆阿勒泰836500;2.眼力健(杭州)制药有限公司,浙江杭州310018 3.浙江省药品检验所,浙江杭州310004;4.杭州海王生物工程有限公司,浙江杭州311101)摘要:目的:探讨在常用中成药降血糖类药品中检测掺杂西药磺酰脲类的分析方法.方法:采用Diamonsil(R)C18柱,以乙腈-0.1%磷酸溶液为流动相,采用梯度洗脱,用二极管阵列检测器检测并对检出的磺酰脲类成分采用质谱检测仪验证.结果:格列吡嗪、格列齐特、格列本脲的最低检测限分别为0.05ng、0.4ng和0.35ng,共检测23批样品,其中有3批样品掺杂了磺酰脲类成分.结论:本方法操作简便,灵敏度高,可作为检测中成药降血糖类药品中掺杂磺酰脲类成分的分析方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208071327_382212_1609970_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208071328_382214_1609970_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208071328_382215_1609970_3.jpg
大家好,我做苯磺隆标样的时候,为什么只出溶剂峰,而没有样品峰呢?条件是按参考文献来的,标准品浓度为0.1,0.5,1.0,2.0,3.0ug/ml,最后还用母液10ug/ml进样也是不出样品峰。

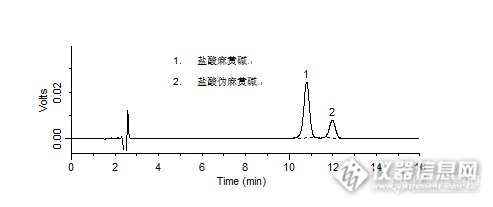
【作者】 谈斐; 蒋学华; 任静; 张岩; 兰轲; 王凌; 田杨;【Author】 TAN Fei,JIANG Xue-hua ,REN Jing,ZHANG Yan,LAN Ke,WANG Ling,TIAN Yang ( West China School of Pharmacy,Sichuan University,Chengdu,Sichuan,610041 P. R. China)【机构】 四川大学华西药学院;【摘要】 目的采用LC-MS/MS法测定人血浆中盐酸伪麻黄碱的血药浓度。方法血浆样品经液-液萃取,结合液-液反相萃取的方法处理,采用DiamonsilTM C18柱(150mm×4.6mm,5μm),以甲醇-10mmol·L-1醋酸铵(冰醋酸调pH4)(20∶80)为流动相,LC-MS/MS用正离子电喷雾离子源(ESI+)、离子选择通道为盐酸伪麻黄碱(m/z166→148)、磷酸可待因内标(m/z300→191)。结果盐酸伪麻黄碱血药浓度5.065~1.013×103ng·ml-1与检测信号具有良好的线性关系(r=0.9986),最低定量浓度为1ng·ml-1。低、次低、中、高浓度的盐酸伪麻黄碱的萃取回收率分别为86.86%±5.8%、95.24%±4.31%、96.91%±1.75%、92.60%±7.79%;内标的萃取回收率为86.24%±3.86%。盐酸伪麻黄碱的日内、日间RSD均小于11.7%。结论所建方法适用于盐酸伪麻黄碱的生物等效性及药物动力学的研究。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301552_380597_2379123_3.jpg
安捷伦5977这段时间样品的丰度下降很多,除了进样口和离子源之外还有什么原因呢?求助

问题:通宣理肺片中麻黄碱的检测:药典要求理论板数按盐酸麻黄碱峰应不低于多少?答案:4000获奖名单:玲儿响叮当(ID:jshbhh)mengzhaocheng(ID:mengzhaocheng)ZHAOGUANGXI(ID:ZHAOGUANGXI)http://ng1.17img.cn/bbsfiles/images/2016/03/201603081524_586278_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603081524_586279_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。通宣理肺片中麻黄碱的检测样品制备 制备方法1. 对照品:取盐酸麻黄碱对照品、盐酸伪麻黄碱对照品适量,精密称定,加0.1 mol/L盐酸溶液制成每1 mL含盐酸麻黄碱30 μg、盐酸伪麻黄碱15 μg的混合溶液,即得。2. 供试品:取装量差异项下的本品,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加水50 mL,密塞,称定重量,超声处理(功率500 W,频率40 kHz)30分钟,放冷,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25 mL,加浓氨试液1 mL,用乙醚振摇提取4次,每次30 mL,合并乙醚液,加5%盐酸乙醇溶液1 mL,摇匀,放置30分钟,蒸干,残渣加水溶解并转移至10 mL量瓶中,加水至刻度,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Platisil ODS 150 x 4.6 mm,5 μm (Cat#:99501)流动相乙腈:0.3%三乙胺的0.02mol/L磷酸二氢钾溶液(用磷酸调节pH值至3.0)=4∶96 流速1.0 mL/min柱温30 ℃检测器UV 210 nm 进样量10 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603081051_586243_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 10.460 581523 37730 10276.215 1.084 -- 2 11.371 238042 14298 10398.086 1.110 2.121 *药典要求理论板数按盐酸麻黄碱峰计算应不低于4000供试品:http://ng1.17img.cn/bbsfiles/images/2016/03/201603081052_586244_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 10.472 993580 64188 10321.811 1.084 -- 2 11.393 343130 20452 10464.593 1.107 2.149 *药典要求理论板数按盐酸麻黄碱峰计算应不低于4000本品种同时使用了SpursilC18色谱柱,在药典规定条件下进行盐酸麻黄碱、盐酸伪麻黄碱的检测,满足药典要求。
本人使用高频熔样机熔片(铝土矿、赤泥),溶剂为偏硼酸锂与四硼酸锂混合熔剂22:12,在熔融过程中不小心被融化的样品粘附在铂黄坩埚外侧,请问除了用热的盐酸浸泡外,还有什么的方法在不损坏坩埚前提下更好的清理?
[size=6][b]西安发现商贩用硫磺熏制生姜(组图)[/b][/size][url=http://www.sina.com.cn/]http://www.sina.com.cn[/url] 2010年09月29日00:25 [url=http://www.cnr.cn/china/gdgg/201009/t20100929_507114574.html]中国广播网[/url] [align=center][img]http://i1.sinaimg.cn/dy/c/p/2010-09-29/1285692049_pY07FO.jpg[/img][/align][align=center]工作人员清点查扣用硫磺熏过的生姜 华商报供图 [/align] [align=center][img]http://i1.sinaimg.cn/dy/c/p/2010-09-29/1285692049_Wua9BE.jpg[/img][/align][align=center]仓库中堆满问题生姜[/align] [align=center][img]http://i3.sinaimg.cn/dy/c/p/2010-09-29/1285692049_GPDQBu.jpg[/img][/align][align=center]左侧颜色比较鲜艳的就是硫磺姜(资料图片)[/align] 中广网西安9月28日消息据中国之声《央广新闻》报道,随着经济的发展,老百姓的菜篮子越来越丰盛,可是这菜篮子里的菜却一而再地出现问题。今年早些时候,江苏南京爆出了使用草酸清洗小龙虾的事件。就在最近,陕西西安又爆出了使用硫磺熏制生姜的“潜规则”。详细情况,我们连线陕西台记者王剑
我现在进行检测枸橼酸离子检测现在的问题是标准出现倒峰,样品峰比溶剂峰还小很多,请问我该怎样消除溶剂峰?谢谢!!我的检测条件是流动相用硫酸,检测器是示差检测器,样品用磺基水杨酸处理,溶剂峰就是磺基水杨酸 。
CNAS举办,辽宁出入境检验检疫局检验检疫技术中心,PTC-T093果酱中柠檬黄日落黄和胭脂红能力验证,这个月24号收到样品了,十个工作日内报结果,有参加的同行吗?大家进来交流下。
农药制剂中硫磺含量的高效液相色谱法测定 汪云强(安徽金泰农药化工有限公司 邮编:231131)摘要 论述了使用C18柱,纯甲醇为流动相,紫外检测器对农药制剂中硫磺含量的反相高效液相色谱测定,方法标准偏差为0.22%,变异系数为0.70%,回收率为98.5%~101.0%之间,对多菌灵• 硫磺、三环唑• 硫磺可湿性粉剂中硫磺含量的测定结果和化学法分析结果基本一致。关键词 硫磺,高效液相色谱,分析1 前言无机硫磺是一种古老的杀菌剂,杀螨剂,由于价格低廉,常和多菌灵、三环唑等复配,加工成多种剂型,如粉剂、可湿性粉剂、悬浮剂等,这些硫磺复配制剂中硫含量的测定,都是采用化学法,步骤繁琐费时。我们经过多次实验,采用反相高效液相色谱法,对多菌灵• 硫磺、三环唑• 硫磺可湿性粉剂中硫磺含量的测定得到较满意的结果。2 实验部分2.1、仪器和试剂色谱仪:LC-10Avp型液相色谱仪(日本岛津),具可变波长紫外检测器色谱柱:VP-ODS 150mm×4.6mm(id)不锈钢柱工作站:JS-3030型 江申通用汉字色谱工作站超声波及0.45μm微孔滤膜甲醇:HPLC级三氯甲烷:AR级硫磺标样:质量分数≥99.0%2.2 色谱操作条件流 动 相:纯甲醇流 量:1.00ml/min检测波长:254nm进样体积:5μl灵敏度:0.08AUFS保留时间:硫磺6.5min2.3 溶液的配制 称取0.03g(精确至0.2mg)标样于50ml容量瓶中,用移液管准确移入 25ml三氯甲烷,超声5min,冷却至室温,摇匀备用。称取含硫磺0.03g(精确至0.2mg)的试样于50ml容量瓶中,用移液管准确移入 25ml三氯甲烷,超声5min,冷却至室温,上清液经0.45μm微孔滤膜过滤后,备用。2.4 测定与计算 在选定的色谱条件下,待仪器基线稳定后,连续注入数针标样溶液,待相邻两针响应值变化小于1.5%时,按标样溶液,试样溶液,试样溶液,标样溶液的顺序进行测定。按峰面积外标法公式计算硫磺的质量百分数。3 结果与讨论3.1 溶剂的选择查有关资料,硫磺能溶于二硫化碳、四氯化碳、三氯甲烷、苯中,微溶于乙醇、甲醇、丙酮中,不溶于水中,二硫化碳沸点较低(46.1℃),挥发性大,三氯甲烷对样品渗透性好,对硫磺溶解快,选用三氯甲烷溶解试样较合适。3.2 检测波长的选择通过752型紫外分光光度计对硫磺在不同波长下的吸收值进行比较,在254nm硫磺有最大吸收。3.3 流动相的选择如果选择流动相ψ(甲醇+水)=95+5 (v/v),硫磺保留时间为13.2min,但如果以纯甲醇作流动相,则基线平稳,复配制剂的其它组分(如多菌灵,三环唑以及溶剂中的三氯甲烷等)均在硫磺前出峰,和硫磺的分离如,且峰形对称,分析时间为6.5min柱压力小等优点。见图1 图13.4 分析方法精密度的测定以含30%硫磺的多• 硫复配制剂为例,在上述色谱条件下,对同一样品中硫含量进行平行测定,方法标准偏差为0.22%,变异系数为0.70%。用已知含量的多• 硫试样,分别加入硫标样,配成溶液,在相同的操作条件下进行测定,计算其回收率在98.5%~101.0%之间。3.5 比较试验选生产中的一个多菌灵• 硫磺样品和一个三环唑• 硫磺样品分别按GB2449-92 测定硫磺的含量和本方法测定硫磺的含量结果如下表:化学法和HPLC法测定硫磺含量比较制 剂化学法HPLC法差值%50%多菌灵• 硫WP30.2230.130.3045%三环唑• 硫WP40.1740.080.22结论:试验结果表明,该方法具有分离效果好,准确度和精密度高,间便快速等特点,是一种理想的分析方法。不妥的地方,敬请各位批评指教
问题:鼻渊通窍颗粒中麻黄碱的检测:盐酸麻黄碱与盐酸伪麻黄碱的分离度是多少呢?答案:2.362幸运奖获得者:吕梁山(ID:shih20j07)dahua1981(ID:dahua1981)梧桐(ID:mengzhou)http://ng1.17img.cn/bbsfiles/images/2015/11/201511251525_575036_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511251525_575037_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511251526_575038_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511251526_575039_1987954_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。鼻渊通窍颗粒中麻黄碱的检测样品制备 制备方法对照品:取盐酸麻黄碱对照品、盐酸伪麻黄碱对照品适量,精密称定,加水制成每1 mL含盐酸麻黄碱15 μg、盐酸伪麻黄碱5 μg的混合溶液,即得。 分析条件 色谱柱Diamonsil C18 150 x 4.6 mm,5 μm (Cat#:99901)流动相乙腈:0.2%磷酸溶液:三乙胺=3.8:96:0.2流速1.0 mL/min柱温30 ℃检测器UV 210 nm进样量20 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2015/11/201511251003_574998_1987954_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 10.792 447325 24164 7959.165 0.957 -- 2 11.976 150547 7676 8532.069 0.948 2.362 *药典要求理论板数按盐酸麻黄碱峰计算应不低于3000,盐酸麻黄碱与盐酸伪麻黄碱的分离度应符合要求。本品种同时使用了Diamonsil C18(2)色谱柱,在药典规定条件下进行麻黄碱的检测,满足药典要求。
1、你用伍丰液相分析什么样品?2、你用的是伍丰那一款型号的液相?3、用了多长时间了?回答每个问题给予2积分奖励。







 我要推广仪器
我要推广仪器
 下载APP
下载APP