XPS各类问题解答及知识详解
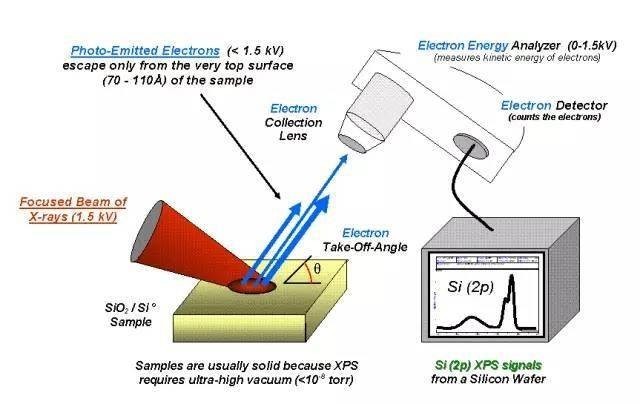
XPS是什么?XPS, 全称为X-ray Photoelectron Spectroscopy(X射线光电子能谱), 早期也被称为ESCA(Electron Spectroscopy for Chemical Analysis),是一种使用电子谱仪测量X-射线光子辐照时样品表面所发射出的光电子和俄歇电子能量分布的方法。XPS可用于定性分析以及半定量分析, 一般从XPS图谱的峰位和峰形获得样品表面元素成分、化学态和分子结构等信息,从峰强可获得样品表面元素含量或浓度。XPS测表面还是体相?XPS是一种典型的表面分析手段。其根本原因在于:尽管X射线可穿透样品很深, 但只有样品近表面一薄层发射出的光电子可逃逸出来。样品的探测深度(d)由电子的逃逸深度(λ, 受X射线波长和样品状态等因素影响)决定,通常,取样深度d = 3λ。对于金属而言λ为0.5-3 nm 无机非金属材料为2-4 nm 有机物和高分子为4-10 nm。XPS定性分析XPS可用于定性分析什么信息?其基本原理是什么?XPS可以定性分析:1) 样品表面元素组成;2) 样品表面元素的化学态和分子结构。A. XPS定性分析元素组成基本原理——光电离作用:当一束光子辐照到样品表面时,光子可以被样品中某一元素的原子轨道上的电子所吸收,使得该电子脱离原子核的束缚,以一定的动能从原子内部发射出来,变成自由的光电子,而原子本身则变成一个激发态的离子。根据爱因斯坦光电发射定律有:Ek =hν- EB式中,Ek为出射的光电子动能;hν为X射线源光子的能量;EB为特定原子轨道上的结合能(不同原子轨道具有不同的结合能)。从式中可以看出,对于特定的单色激发源和特定的原子轨道,其光电子的能量是特征的。当固定激发源能量时,其光电子的能量仅与元素的种类和所电离激发的原子轨道有关。因此,我们可以根据光电子的结合能定性分析物质的元素种类。B.XPS定性分析元素的化学态与分子结构基本原理:原子因所处化学环境不同,其内壳层电子结合能会发生变化,这种变化在谱图上表现为谱峰的位移(化学位移)。这种化学环境的不同可以是与原子相结合的元素种类或者数量不同,也可能是原子具有不同的化学价态。一般规律:1) 氧化作用使内层电子结合能上升,氧化中失电子愈多,上升幅度愈大 2)还原作用使内层电子结合能下降,还原中得电子愈多,下降幅度愈大 3) 对于给定价壳层结构的原子,所有内层电子结合能的位移几乎相同.XPS的定量分析A. 基本原理经X射线辐照后,从样品表面出射的光电子的强度(I,指特征峰的峰面积)与样品中该原子的浓度(n)有线性关系,因此可以利用它进行元素的半定量分析。简单的可以表示为:I = n*S, S称为灵敏度因子(有经验标准常数可查,但有时需校正)。对于对某一固体试样中两个元素i和j, 如已知它们的灵敏度因子Si和Sj,并测出各自特定谱线强度Ii和Ij,则它们的原子浓度之比为:ni:nj=(Ii/Si):(Ij/Sj),因此可以求得相对含量。B. 为什么XPS是一种半定量分析手段?鉴于光电子的强度不仅与原子的浓度有关,还与光电子的平均自由程、样品的表面光洁度,元素所处的化学状态,X射线源强度以及仪器的状态有关。因此,XPS技术一般不能给出所分析元素的绝对含量,仅能提供各元素的相对含量。谱图处理(1)寻找主要的光电子峰——鉴定元素(H和He除外,探测深度约2-10nm);(2)伴峰的识别——帮助解析谱图,为原子中电子结构的研究提供重要信息;(3)本底扣除(直线法和非直线法-Shirley法等);(4) 峰的拟合(参数:峰位、峰高、半峰宽、G/L比等)。• 本底--韧致辐射(非弹性散射的一次和二次电子产生),高结合能的背底电子多,随结合能的增高呈逐渐上升的趋势。• 光电子谱线--每一种元素都有自己特征的光电子线,它是元素定性分析的主要依据。谱图中强度最大、峰宽最小、对称性最好的谱峰,由未经非弹性散射的光电子形成,称为XPS的主谱线。实例说明:上图中,对于Zn元素而言,Zn 2p强度最大、峰宽最小,对称性最好,是Zn元素的主谱线。而除了主谱线Zn 2p之外,其实还有Zn 3s、Zn 3p和Zn 3d等其它谱线,这是因为Zn元素有多种内层电子,因而可以产生多种In XPS信号。标定顺序(1)利用污染碳的C1s或其他方法扣除荷电效应,常见方法是以测量值和参考值(284.8eV)之差作为荷电校正值(Δ)来矫正谱中其他元素的结合能,整个过程中XPS谱图强度不变;(2)鉴别总是存在的元素谱线,如C、O的谱线;(3)鉴别样品中主要元素的强谱线和有关的次强谱线;(4)鉴别剩余的弱谱线假设它们是未知元素的最强谱线。自旋-轨道分裂(SOS)由于电子的轨道运动和自旋运动发生耦合后使轨道能级发生分裂。对于l>0的内壳层来说,用内量子数j(j=|l± ms|)表示自旋轨道分裂。即若l=0 则j=1/2;若l=1则j=1/2或3/2。除了s能级是单峰外,p、d、f都分裂为双峰。实例说明:上图所示为ZnO的XPS谱图,图中Zn 2p裂分成2p3/2和2p1/2双峰。重要意义:对于某一特定价态的元素而言,其p、 d、 f 等双峰谱线的双峰间距及峰高比一般为一定值。p峰的强度比为1:2;d线为2:3;f线为3:4。对于p峰,特别是4p线,其强度比可能小于1:2。高分辨谱定性分析元素的价态主要看两个点(1)可以对照标准谱图值(NIST数据库或者文献值)来确定谱线的化合态;(2)对于p,d,f 等具有双峰谱线的(自旋裂分),双峰间距也是判断元素化学状态的一个重要指标。实用XPS定量分析方法可以概括为标样法、元素灵敏度因子法和一级原理模型。目前XPS定量分析多采用元素灵敏度因子法,该方法利用特定元素谱线强度作参考标准,测得其它元素相对谱线强度,求得各元素的相对含量。定量分析方法步骤(1)扣除背景(Shirley,Linear,Touggard);(2)测量峰面积(必要时进行峰拟合);(3)应用灵敏度因子;(4)计算原子浓度。实例说明:上表为三种ZnO的Zn和O元素的相对含量,其中O 1s峰形如图是不对称的,说明是由不止一个峰组成的。根据谱峰形状,结合文献对O 1s进行峰拟合,分别是530 eV左右的ZnO晶格O的峰(OL)、531.7 eV左右的羟基O(Oα)的峰和533 eV的吸附O(Oβ)的峰。根据峰面积,结合各元素灵敏度因子,计算得到的元素比例如表所示。XPS的分析方法1. XPS定性分析元素组成的范围?为什么?XPS常用Al Kα或者Mg Kα X射线为激发源,能检测周期表中除氢、氦以外的所有元素,一般检测限为0.1%(原子百分数)。XPS之所以无法检测H, He是因为:1) H和He的光电离界面小,信号太弱;2) H1s电子很容易转移,在大多数情况下会转移到其他原子附近,检测起来非常困难 3) H和He没有内层电子,其外层电子用于成键,H以原子核形式存在。所以用X射线去激发时,没有光电子可以被激发出来。2. XPS定性分析的具体方法A. 化合物中元素种类的分析——全谱分析(1) 什么时候需要进行全谱分析(XPS survey)?全谱分析的目的是什么?全谱分析一般用来说明样品中是否存在某种元素。比较极端的,对于某一化学成分完全未知的样品,可以通过XPS全谱分析来确定样品中含有哪些元素(H和He除外)。全谱分析主要看峰的有无,进而确定是否存在该元素。(2)全谱分析有何不足之处?全谱分析所得到的信号比较粗糙,只是对元素进行粗略的扫描,确定元素有无以及大致位置。对于含量较低的元素而言,信噪比很差,不能得到非常精细的谱图。通常,全谱分析只能得到表面组成信息,得不到准确的元素化学态和分子结构信息等。(3)XPS全谱分析与EDS有何异同?a. EDS与XPS的相同点:两者均可以用于元素的定性和定量检测。b. EDS与XPS的不同点:1) 基本原理不一样: 简单来说,XPS是用X射线打出电子,检测的是电子;EDS则是用电子打出X射线,检测的是X射线。2) EDS只能检测元素的组成与含量,不能测定元素的价态,且EDS的检测限较高(含量>2%),即其灵敏度较低。而XPS既可以测定表面元素和含量,又可以测定表其价态。XPS的灵敏度更高,最低检测浓度>0.1%。3) 用法不一样:EDS常与SEM,TEM联用,可以对样品进行点扫,线扫,面扫等,能够比较方便地知道样品的表面(和SEM联用)或者体相(和TEM联用)的元素分布情况;而XPS则一般独立使用,对样品表面信息进行检测,可以判定元素的组成,化学态,分子结构信息等。B. 化合物中化学态与结构分析——窄区扫描(高分辨谱)(1) 什么叫荷电校正?为什么需要进行荷电校正?当用XPS测量绝缘体或者半导体时,由于光电子的连续发射而得不到电子补充,使得样品表面出现电子亏损,这种现象称为“荷电效应”。荷电效应将使样品表面出现一稳定的电势Vs,对电子的逃离有一定束缚作用。因此荷电效应将引起能量的位移,使得测量的结合能偏离真实值,造成测试结果的偏差。在用XPS测量绝缘体或者半导体时,需要对荷电效应所引起的偏差进行校正(荷电校正的目的),称之为“荷电校正”。(2)如何进行荷电校正?最常用的,人们一般采用外来污染碳的C1s作为基准峰来进行校准。以测量值和参考值(284.8 eV)之差作为荷电校正值(Δ)来矫正谱中其他元素的结合能。具体操作:1) 求取荷电校正值:C单质的标准峰位(一般采用284.8 eV)-实际测得的C单质峰位=荷电校正值Δ;2)采用荷电校正值对其他谱图进行校正:将要分析元素的XPS图谱的结合能加上Δ,即得到校正后的峰位(整个过程中XPS谱图强度不变)。将校正后的峰位和强度作图得到的就是校正后的XPS谱图。(3)如何通过高分辨谱判定样品中某种元素的价态?高分辨谱定性分析元素的价态主要看两个点:1)可以对照标准谱图值(NIST数据库或者文献值)来确定谱线的化合态;2)对于p,d,f等具有双峰谱线的(自旋裂分),双峰间距也是判断元素化学状态的一个重要指标。(4)高分辨谱的其他常见用途实际上,多数情况下,人们关心的不仅仅是表面某个元素呈几价,更多的是对比处理前后样品表面元素的化学位移变化,通过这种位移的变化来说明样品的表面化学状态或者是样品表面元素之间的电子相互作用。一般,某种元素失去电子,其结合能会向高场方向偏移,某种元素得到电子,其结合能会向低场方向偏移,对于给定价壳层结构的原子,所有内层电子结合能的位移几乎相同. 这种电子的偏移偏向可以给出元素之间电子相互作用的关系。实例说明:下图所示是PtPd形成合金后其表面电子结构的变化,从图中可以看出,形成Pt1Pd3之后,Pd3d向低场偏移,Pt4f向高场偏移,说明Pd得到电子,Pt失去电子,也就是说形成合金后,Pt上的电子部分转移给Pd。PtPd的这种电子转移也是其形成合金的一个证据。XPS谱图中有哪些重要的谱线结构?XPS谱图一般包括光电子谱线,卫星峰(伴峰),俄歇电子谱线,自旋-轨道分裂(SOS)等1. 光电子谱线:每一种元素都有自己特征的光电子线,它是元素定性分析的主要依据。谱图中强度最大、峰宽最小、对称性最好的谱峰,称为XPS的主谱线。实例说明:上图中,对于In元素而言,In 3d强度最大、峰宽最小,对称性最好,是In元素的主谱线。而除了主谱线In 3d之外,其实还有In 4d, In 3p等其它谱线,这是因为In元素有多种内层电子,因而可以产生多种In XPS信号。2. 卫星峰(伴峰):常规X射线源(Al/Mg Kα1,2)并非是单色的,而是还存在一些能量略高的小伴线(Kα3,4,5和Kβ等),所以导致XPS中,除Kα1,2所激发的主谱外,还有一些小的伴峰。实例说明:上图为Mg阳极X射线激发的C1s主峰(α1,2)及伴峰(α3,4,5和β)。从图中可以看出,主峰的强度比伴峰要强很多。3. 俄歇电子谱线:电子电离后,芯能级出现空位,弛豫过程中若使另一电子激发成为自由电子,该电子即为俄歇电子。俄歇电子谱线总是伴随着XPS,但具有比XPS更宽更复杂的结构,多以谱线群的方式出现。特征:其动能与入射光hν无关。实例说明:上图中O KLL, C KLL即为O和C的俄歇电子谱线,从图中可以看到O KLL其实有三组峰,最左边的为起始空穴的电子层,中间的是填补起始空穴的电子所属的电子层,右边的是发射俄歇电子的电子层。4. 自旋-轨道分裂(SOS):由于电子的轨道运动和自旋运动发生耦合后使轨道能级发生分裂。对于l>0的内壳层来说,用内量子数j(j=|l±ms|)表示自旋轨道分裂。即若l=0 则j=1/2;若l=1则j=1/2或3/2。除s亚壳层不发生分裂外,其余亚壳层都将分裂成两个峰。实例说明:上图所示为PbO的XPS谱图,图中Pb 4f裂分成4f5/2和4f7/2两个峰。重要意义:对于某一特定价态的元素而言,其p, d, f 等双峰谱线的双峰间距及峰高比一般为一定值。p峰的强度比为1:2;d线为2:3;f线为3:4。对于p峰,特别是4p线,其强度比可能小于1:2。双峰间距也是判断元素化学状态的一个重要指标。5. 鬼峰:有时,由于X射源的阳极可能不纯或被污染,则产生的X射线不纯。因非阳极材料X射线所激发出的光电子谱线被称为“鬼峰”。





























 我要推广仪器
我要推广仪器
 下载APP
下载APP