GB2763中关于亚砜磷的限值要求,标准中指出残留物为亚砜磷 砜吸磷 甲基内吸磷之和,以亚砜磷表示,不知道亚砜磷 砜吸磷 甲基内吸磷这三者之间有什么关系,是可以相互转化,还是同分异构体?
我用的是紫外可见光分光光度计 型号是WFZ800-D3B在测定硝态氮时发现吸收峰出现在200nm,测定全磷时吸收峰在880-900nm比较大,700nm很小,请教是什么原因?多谢各位!
甲拌磷、甲拌磷砜、甲拌磷亚砜在气相出峰如何,三个峰会重合吗?检测条件如何,有图谱发上来,会有积分奖励。
用气相可以做甲拌磷砜与甲拌磷亚砜吗?上级下的任务是检测有机磷农药残留,其中有甲拌磷、甲拌磷砜与甲拌磷亚砜,甲拌磷出峰很好,但这两种能出峰吗?标液还在购买中,希望做过的说说出峰情况。
FPD检测器,DB1701(30*0.25*0.25),以前做甲拌磷砜与甲拌磷亚砜,可以分离,两峰相差0.5min,买了新DB1701柱,老化后进样,砜与亚砜两峰完全重合,是标液标错了,还是新柱有问题,但其它20种农药出峰都正常,有人遇见过这种情况吗?
甲拌磷砜GB23200.8-2016,甲拌磷亚砜有国标或农业部的检测方法么,求标准号
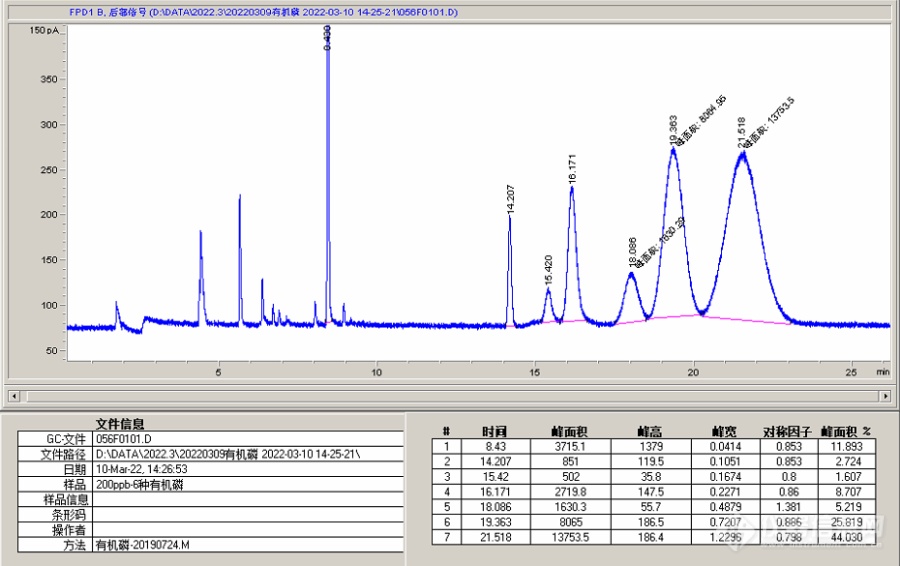
仪器是安捷伦7890A,PFD检测器,色谱柱是DB-1701,30m×0.25mm×0.25μm,柱流量1.5ml/min,进样口220℃,不分流进样,检测器氢气70ml/min,空气100ml/min,尾吹60ml/min,升温程序参考的之前版上高手发过的,80℃保持1min,以20℃/min升温至130℃,再以5℃/min升温至200℃,最后以15℃/min升温至250℃,保持11min。分析6种有机磷混标,包括敌敌畏、乐果、内吸磷、甲基对硫磷、马拉硫磷、对硫磷,除了敌敌畏和内吸磷以外,其他物质峰形都很难看,峰宽矮胖,马拉硫磷与对硫磷也分不开(我在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上HP-5MS柱子同样升温程序可以完美分开这两种物质)。也试过其他升温程序和柱流量(1~2mL/min),包括之前换过其他HP-5等柱子,始终出现类似峰形无法分开马拉硫磷与对硫磷,怀疑是否这台[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]的FPD检测器有问题?请教各位高手指点[img=,690,433]https://ng1.17img.cn/bbsfiles/images/2022/03/202203292320019341_7010_1916553_3.png!w690x433.jpg[/img]
检测好几种橙子里在烯酰吗啉处都有干扰峰,对烯酰吗啉的双峰有一定干扰,不过还好不影响定量,准备农业部农残考核的伙伴们有遇到这种情况的吗?还是说所有橙子里都在烯酰吗啉这个地方有干扰?
农业部的能力验证又检出甲拌磷亚砜,按照刘主任的要求需要将其换算成甲拌磷来计算最终的结果。两者之间是怎样的换算关系?比如检出甲拌磷0.050,甲拌磷亚砜0.004
[color=#444444]有网友问:我看之前论坛您发过水胺硫磷不出峰的问题,现在我也遇到这种困扰。做6种农药单标保留时间确认,毒死蜱、甲胺磷、甲拌磷、甲基异柳磷、氧乐果、水胺硫磷,最后走的水胺硫磷,浓度统一为0.2μg/mL,发现水胺硫磷响应值比其他低几十倍,标液出了几个响应值差不多的峰,但都不是前述几个的残留,您分析是什么问题呢[/color]
参照NY/T 761-2008的做法,发现硫环磷、甲基硫环磷、蝇毒磷、保棉磷标液在FPD检测器上不出峰,0.1和1.0ug/mL的标样都试过了,DB-1701柱,时间设的也够久了,最后250摄氏度走了22min。用GC-MS硫环磷、蝇毒磷、保棉磷出峰正常,甲基硫环磷响应很低。请各位给分析分析,谢谢。

亚胺硫磷与甲拌磷亚砜多大浓度可以出峰?这两种标液要配到1.0ug/mL才能出峰,这是为什么?其它农药标液出峰正常。[img=,690,137]http://ng1.17img.cn/bbsfiles/images/2017/05/201705241852_01_1645480_3.jpg[/img]
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]三重四级杆跑了甲拌磷和甲拌磷亚砜 为什么出峰时间是重叠的 甲拌磷和甲拌磷亚砜应该离的挺远啊 求帮助
想问下各位大神,用30米的1701的色谱柱和单杆[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]测甲拌磷亚砜和甲拌磷砜有什么升温程序可以把两个峰分开吗
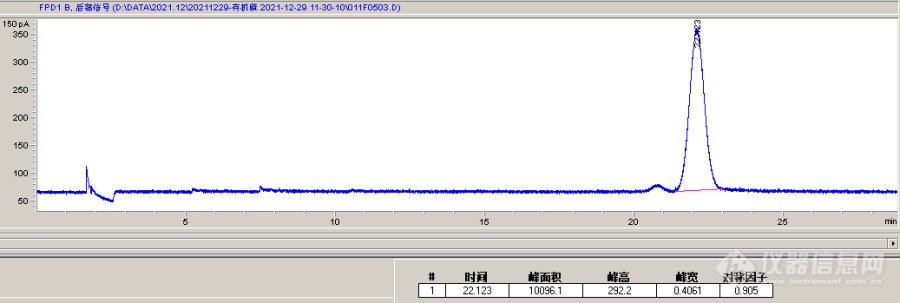
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]为安捷伦7890A,色谱柱为30米长、0.25mm口径、膜厚0.25μm的HP-5MS柱,进样口230℃,不分流进样,柱流量1.0 mL/min,程序升温条件为80℃保持1min,20℃/min升温至130℃,5℃/min升温至200℃,15℃/min升温至250℃保持8min,检测器250℃,氢气流量75mL/min,空气流量100mL/min,尾吹流量30mL/min。分析丙酮标液中的敌敌畏、敌百虫、乐果、甲基对硫磷、马拉硫磷、对硫磷共6种有机磷,进样浓度 2 mg/L,前面的物质出峰都挺高的,峰形也还可以,但马拉硫磷和对硫磷出峰峰形差,峰宽很宽,因此两种物质出峰基本重叠无法分开,试了好多程序升温条件都难以改善,请教各位老师这种情况是什么原因?如何解决?我也试过换一根新的0.32mm的HP-5柱子,情况类似,看论坛里多数人推荐DB-1701的柱子,可是我们实验室只有一根1.0μm膜厚的1701柱,换上去测试出峰太慢了,敌敌畏要12min以后才出峰,马拉硫磷和对硫磷在260℃保持20min后都不见出峰[img]https://simg.instrument.com.cn/bbs/images/brow/em49.gif[/img]图中为2mg/L马拉硫磷单标进样的色谱图,2mg/L对硫磷单标进样的色谱图出峰时间、峰形基本与之类似。[img=,690,232]https://ng1.17img.cn/bbsfiles/images/2021/12/202112301058143478_7214_1916553_3.png!w690x232.jpg[/img]
用气质走了一下甲拌磷砜和亚砜,发现两个的质谱图基本一样,出峰时间也差不多重叠,那么混标怎么做呢。我是做农残的,如果不能分开,怎么确定目标物,哪位大神有好办法或者能分开的仪器参数,升温程序。
配制甲拌磷亚砜1.0ug/mL,出现两个峰,前一个峰很小,而且与甲拌磷出峰时间重合,那么甲拌磷亚砜出几个峰,与甲拌磷重合的小峰也是甲拌磷亚砜的吗?
安捷伦7890A-5977B检测甲胺磷和乙酰甲胺磷,1ppm基本不出峰,程序升温其他农残成分都能出峰,用的HP-5柱子,在预测保留时间有个特别小的峰应该是,感觉就是分解了或吸附了,怎么办?求助
实验室参数甲拌磷,如何能力验证要求测甲拌磷砜和甲拌磷亚砜,购买甲拌磷标物用761[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]可以测吗?

东西(IC2800),走淋洗液,基线有剧齿峰,很明显!! 详细过程:第一针进样品,在水峰前有个大的峰,几乎看不到水峰,没有明显的剧齿峰;第二针进标液,同样水峰前面有个莫名其妙的大峰,水峰不明显;第三针重进上面的标液, 水峰前面没有 “莫名其妙的大峰”,水峰显现,标液基本有形状, 不过基线伴有轻微的剧齿峰;第四针进另一个高浓度的标液,现象同上一样;第五针进了一个样品,图的情况不记得了。第六针进又进标 液出问题了, 基线剧齿峰非常明显, 而且整个峰 往下降。 结束重新开启,走淋洗液,剧齿峰同样地明显,整个峰又向上抬起! 整个就是乱糟糟地感觉!!http://ng1.17img.cn/bbsfiles/images/2011/12/201112021110_334864_1639541_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/12/201112021110_334865_1639541_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/12/201112021110_334866_1639541_3.jpg
最近在做农残扩项,在做到烯酰吗啉的时候MRM出的是双峰,标样一开始买的农科院环保所的,后来买的DR.E的,都是一样的情况。流动相是甲酸水乙腈,初始流动相定容的。[img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806071421523441_3917_3309755_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806071421524367_1575_3309755_3.jpeg[/img]
海藻糖用多少浓度淋洗液出峰?多少流速?柱子是PA1 PA20 我们的淋洗液是200mmol的氢氧化钠和500mmol的醋酸钠
特丁硫磷和特丁硫磷亚砜的离子对是多少?有谁做过,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]条件是什么?
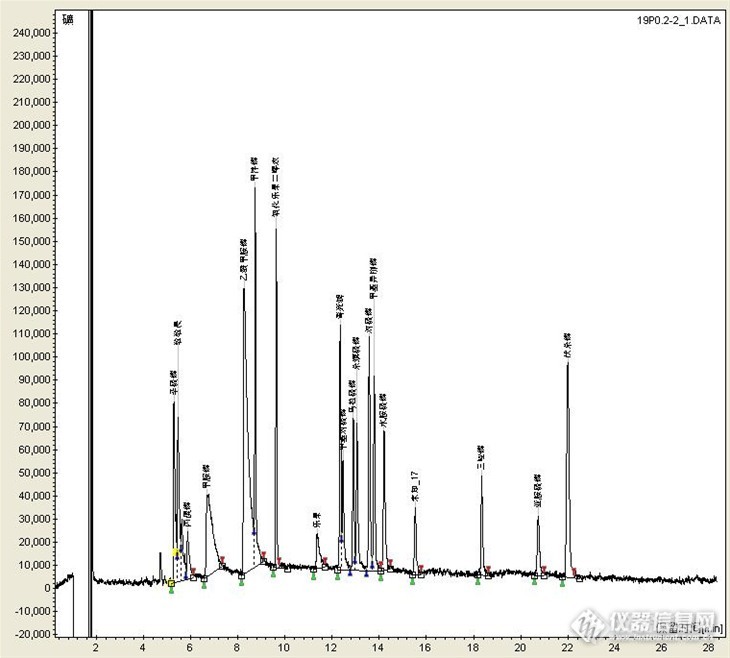
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231601_441223_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
我用GC-FPD检测内吸磷信号很强,相同的GC条件在GC-MS上内吸磷没有峰了,这是怎么回事呢?难道内吸磷没有离子碎片?请大虾们指教。谢谢了!
限量标准上说的是合计以甲拌磷、涕灭威……计,那么最后数据处理计算时该怎么计算,峰面积加和还是分子量换算?不是很理解,请各位大佬指点。还有根据标准上保留时间及相对离子丰度该如何判定样品是否检出,是否在仪器离群值那里设定后,离群就可以判断其未检出还是需要自己根据实际情况再做判断?
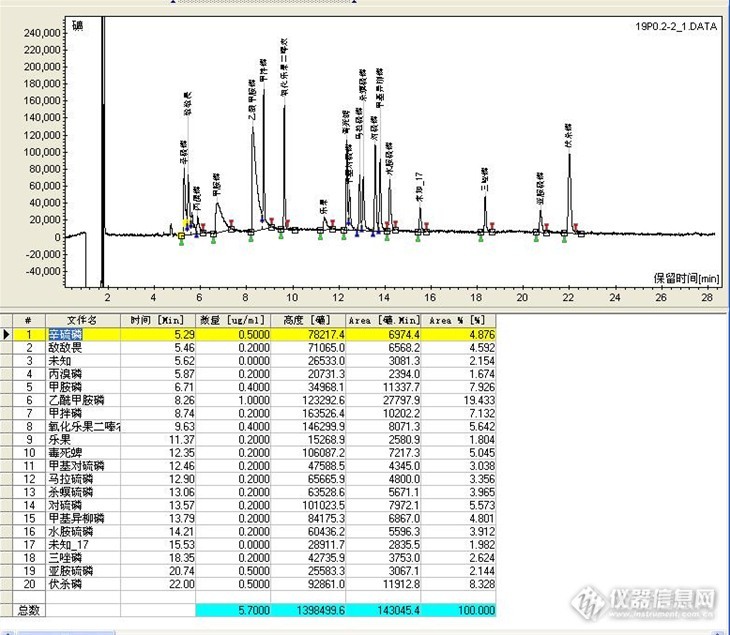
用FPD检测器,DB1701色谱柱做乙酰甲胺磷时,容易被衬管吸附,用高惰性衬管出峰,但峰有拖尾现象,那么有没有人用别的型号的色谱柱做乙酰甲胺磷,检出限低而出峰尖锐?http://ng1.17img.cn/bbsfiles/images/2013/05/201305231603_441225_1645480_3.jpg这是DB1701(30*0.25*0.25)色谱柱的乙酰甲胺磷出峰情况。
仪器: Agilent 7890A/5975C 柱子DB-5MS问题: GC/MS做有机磷农药 甲醇标样:5ppm不出峰 100ppm出峰 做SVOC标样5ppm峰形都很好怀疑过玻璃棉吸附,但是问过安捷伦工程师说没有吸附那么厉害;换过进样垫,照样不出峰,请问各位是啥问题呢?看过论坛上 解决方法:将柱子进样口端拆下,截去一段;用甲醇清洗进样口;换玻璃棉;重新安装接口处柱子,使伸入长度在1mm左右。 不知道是否适合我们呢,请各位帮忙,谢谢!
甲基对硫磷用程序升温时出峰很好,它与19种有机磷一起出峰,用DB1701柱,可是只有它一个时用恒温出峰很难看,恒温是190度保持20min,其它条件一样,只是程序升温与恒温的区别会影响峰形与峰面积吗?
衬管容易吸附亚胺硫磷吗?刚换上的新衬管,亚胺硫磷0.2ug/mL就出峰,可进一批样品后,再进亚胺硫磷,不出峰,一直配到5.0ug/mL才出一点峰,是衬管吸附太厉害吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP