最近我们实验室正在进行国家土壤盲样考核,要检出多环芳烃,质控样中干扰物质太多,如何除去其他杂质?
大家好,想请教下问如何除去多环芳烃(非极性)中的少量脂肪烃,谢谢。
色谱非芳烃的计算
各位大神,因为实验要求,要用ESI[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]连用来测非取代多环芳烃,但是选什么类型的色谱柱呢 会不会正负模式 质朴都没信号呢 求指教
提取:用正己烷提取净化的时候:用的是Agilent的C18固相萃取柱,500mg的 活化的时候用5ml二氯甲烷+5ml甲醇+5ml水 然后上样,淋洗的时候用的是5ml正己烷,最后洗脱用的是5ml二氯甲烷这样做出来的样品,谱图上还是检测到很多很强的杂质峰,请问这是为什么呀?是哪里不对吗?另一个问题是,用正己烷来做淋洗溶液,会不会把样液中的多环芳烃洗脱出来呀?我对用正己烷做淋洗液这个做法存在疑惑?不知可不可以这么做?
求助!请问下多环芳烃标液怎么配置?还有菲的标液怎么配置标液,需要哪些条件?
第一次做多环芳烃,用的是HP-5的柱子。前两天做邻苯都可以出峰的,进了一针2ug/mL还有8ug/mL16种PAHs混标标液目标峰一点峰都没出,就出了几个正己烷中的杂质峰,衬管用的是石英棉的分流衬管,用的是不分流进样。仪器进样口温度还有程升温度都调了很多次还是出不来。有大神可以帮帮忙不。。气质方面完全新手一枚。。
最近在用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]测定某注射液中的多环芳烃杂质,但是在分析空白溶剂二氯甲烷的时候总是出现萘的残留,清洗进样针,更换衬管,进样隔垫,均没有改善,但是走空针的时候没有残留,不知道到是什么原因,求帮助

[align=center][b]流动相杂质捕集小柱(Ghost-Buster Column)在多环芳烃(PAHs)测定中的应用[/b][/align][align=left][b]摘要[/b]: 高效液相色谱法(HPLC)测定多环芳烃(PAHs),连接月旭公司流动相杂质捕集小柱(Ghost-BusterColumn)后,空白溶剂(色谱纯乙腈)和PAHs标准品(10ppb)的进样色谱图相比较没有添加时,流动相梯度基线漂移明显改善,基线更平滑,鬼峰(Ghostpeak)明显减少,更利于多环芳烃(PAHs)的定性定量。[/align][align=left][b]Abstract: [/b]In thedetermination of PAHs with HPLC, after adding GhostBuster Column, it is shownthrough chromatogram of blank solvent and PAHs standard that the baseline driftis improved and Ghost peak reduce compared with no adding, which is more conduciveto qualitative and quantitative of PAHs.[/align][align=left][b]关键词: [/b]高效液相色谱法;多环芳烃;流动相杂质捕集小柱,鬼峰[/align][align=left][b]Key words: [/b]HPLC;PAHs;Ghost-BusterColumn;Ghostpeak[/align][align=left][b]0 引言[/b][/align][align=left] 多环芳烃(Polycyclic AromaticHydrocarbons, PAHs)是指分子结构中含有两个或两个以上并环苯环的烃类化合物,广泛存在于人类生活的自然环境如大气、水体、土壤中,同时存在于作物和食品中。迄今已发现200多种PAHs,其中有相当部分具有致癌性,因而其检测备受关注。多环芳烃(PAHs)的检测方法随科技发展在不断进步,从开始的柱层析、纸色谱、薄层色谱(TLC)和凝胶渗透色谱(GPC)发展到如今的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](GC)、高效液相色谱(HPLC),还有紫外吸收光谱(UV)、发射光谱(包括荧光、磷光和低温发光等)、质谱分析、核磁共振和红外光谱,以及各种分析方法之间的联用技术等。目前较为常用的是高效液相色谱法(HPLC)[sup][/sup]。 液相色谱法测定18种多环芳烃的过程中,特别是在梯度洗脱过程中容易产生时有时无的色谱峰,我们俗称鬼峰(Ghostpeak)。荧光检测器灵敏度高,检测过程中鬼峰严重干扰目标峰,影响PAHs定量,月旭公司流动相杂质捕集小柱(Ghost-BusterColumn)能有效解决这一问题,未来或将成为液相色谱运行梯度分析时的标配。[/align][align=left][b]1 实验部分[/b][/align][align=left][b]1.1 实验材料和仪器[/b][/align][align=left] 仪器和耗材包括:高效液相色谱仪LC-20AD(带荧光检测器RF-20A),日本岛津仪器公司;Ultimate[sup][/sup]PAH(5 um, 4.6×250 mm),月旭科技有限公司;Buster-GhostColumn流动相杂质捕集小柱(4.6×50mm),月旭科技有限公司;超纯水机,成都优普公司;色谱试剂,美国斯百全公司;PAHs标准品(18种),国家标准物质中心。[/align][align=left][b]1.2 色谱条件[/b][/align][align=left] 流动相: 流动相A:水 流动相B:乙腈 流速1.5mL/min,[/align][align=left] 柱温35℃,[/align][align=left] 进样量20μL 表1列出了溶剂洗脱程序(梯度洗脱程序的条件应根据选用的仪器进行调整)。[/align][align=center]表1流动相梯度洗脱程序[/align] [table][tr][td][align=center] [/align][align=center]时间(min)[/align][align=center] [/align][/td][td][align=center] [/align][align=center]流动相A(%)[/align][align=center] [/align][/td][td][align=center] [/align][align=center]流动相B(%)[/align][align=center] [/align][/td][/tr][tr][td][align=center] [/align][align=center]0[/align][align=center] [/align][/td][td][align=center] [/align][align=center]50[/align][align=center] [/align][/td][td][align=center] [/align][align=center]50[/align][align=center] [/align][/td][/tr][tr][td][align=center] [/align][align=center]30[/align][align=center] [/align][/td][td][align=center] [/align][align=center]10[/align][align=center] [/align][/td][td][align=center] [/align][align=center]90[/align][align=center] [/align][/td][/tr][tr][td][align=center] [/align][align=center]40[/align][align=center] [/align][/td][td][align=center] [/align][align=center]10[/align][align=center] [/align][/td][td][align=center] [/align][align=center]90[/align][align=center] [/align][/td][/tr][tr][td][align=center] [/align][align=center]45[/align][align=center] [/align][/td][td][align=center] [/align][align=center]50[/align][align=center] [/align][/td][td][align=center] [/align][align=center]50[/align][align=center] [/align][/td][/tr][/table] 18种PAHs中,苊烯无荧光吸收,而剩余的17种有不同的最大激发和发射波长[sup][/sup],综合考虑后选取了3个,在保证定性定量的前提下简化测试过程。因仪器及实验条件不同,所以检测波长时间段可以根据实际出峰情况进行设置,以保证17种PAHs完全出峰。表2列出了三个时间段内的荧光检测波长及所测物质。[align=center]表2荧光检测波长随梯度变化的检测方案[/align] [table][tr][td] [align=center]时间(min)[/align] [/td][td] [align=center]检测波长[/align] [/td][td] [align=center]所测物质[/align] [/td][/tr][tr][td] [align=center]0-19[/align] [/td][td] [align=center]Ex=270nm,Em=324nm[/align] [/td][td] [align=center]萘、苊烯(无荧光吸收)、1-甲基萘、2-甲基萘、苊、芴[/align] [/td][/tr][tr][td] [align=center]19-41.05[/align] [/td][td] [align=center]Ex=245nm,Em=425nm[/align] [/td][td] [align=center]菲、蒽、荧蒽、芘、苯并(a)蒽、屈、苯并(b)荧蒽、苯并(k)荧蒽、苯并(a)芘、二苯并(a,h)蒽、苯并(g,h,i)苝[/align] [/td][/tr][tr][td] [align=center]41.05-45[/align] [/td][td] [align=center]Ex=274nm,Em=507nm[/align] [/td][td] [align=center]茚并(1,2,3-cd)芘[/align] [/td][/tr][/table][b]2 结果与讨论[/b][align=left][b]2.1加和不加补集柱时进样空白溶剂时图谱(进样20μL色谱纯乙腈)[/b] [/align][align=left]加和不加补集柱时进样空白溶剂时测试结果,图1a为不加捕集柱进样20 μL色谱纯乙腈时的色谱图,图1b为加捕集柱进样20 μL色谱纯乙腈时的色谱图,图2为加和不加捕集柱进样20 μL色谱纯乙腈时的叠加对比图。[/align][align=center][img=,610,273]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240928_01_2451449_3.png[/img][/align][align=left][/align][align=center][img=,607,274]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240928_02_2451449_3.png[/img][/align][align=center]图1进样20 μL色谱纯乙腈时的色谱图[/align][align=center](a 不加捕集柱的测试结果b. 加捕集柱的测试结果)[/align][align=center][img=,609,194]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240929_01_2451449_3.png[/img][/align][align=center]图2 加和不加捕集柱进样20 μL色谱纯乙腈时的叠加对比图[/align][align=left] 黑色图谱代表不加捕集柱,红色谱图代表加捕集柱。由图2可知,加捕集柱能明显消除鬼峰,基线相比不加捕集柱时更平滑,基线漂移得到明显改善。[/align][align=left][/align][align=left][b]2.2 加和不加捕集柱时进样PAHs时图谱(进样体积20 μL,浓度10 ppb)[/b][/align][align=left] 图3a为不加捕集柱PAHs进样色谱图(进样体积20 μL,浓度10 ppb),图3b为加捕集柱PAHs进样色谱图(进样体积20 μL,浓度10 ppb),图4为加和不加捕集柱PAHs标样叠加对比图。[/align][align=center][img=,618,273]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240931_01_2451449_3.png[/img][/align][align=center][img=,619,268]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240932_01_2451449_3.png[/img][/align][align=center]图3进样20 μL浓度10ppb PAHs时的色谱图[/align][align=center](a 不加捕集柱的测试结果b. 加捕集柱的测试结果)[/align][align=center][img=,594,267]http://ng1.17img.cn/bbsfiles/images/2017/08/201708240933_01_2451449_3.png[/img][/align][align=center]图4为加和不加捕集柱PAHs标样叠加对比图[/align][align=left] 红色图谱代表不加捕集柱,黑色谱图代表加捕集柱。图4可知,加捕集柱能明显消除鬼峰,基线相比不加捕集柱的图也更平滑,更利于PAHs的定性定量。同时,加捕集柱后保留时间有微小位移,原因是捕集柱的加入增加了一段小的管路体积,但不影响最终定量分析。[/align][align=left][b][/b][/align][align=left][b]3.结论[/b][/align][align=left][/align][align=left] 因为18种PAHs中苊烯没有荧光吸收,所以实际测得17种;加和不加补集柱,测得的17种多环芳烃分离度均达到要求;月旭杂质捕集小柱(Ghost-BusterColumn)添加后能有效去除鬼峰对目标物的干扰,运行梯度时基线漂移明显改善,更利于PAHs定性定量分析,未来或将成为液相色谱运行梯度分析时的标配。[/align][align=left][b][/b][/align][align=left][b]4 参考文献[/b][/align][align=left][b][/b][/align][align=left]美国国家环保局. US EPA Method 610[/align][align=left]中华人民共和国国家质量监督检验检疫总局. GB/T24893-2010/ISO 15753:2006[/align]
[font=宋体]链接:[/font]https://bbs.instrument.com.cn/topic/2856357问题描述:[font=宋体]做沉积物多环芳烃,用二氯甲烷索氏提取[/font]48[font=宋体]小时,加了铜片,发现有的样品提取液是无色的,有的是黄色,请问是怎么回事,是铜片不够了,已经加了很多了。[/font]解答:[font=宋体]在进行土壤或沉积物等样品有机分析过程中,应加入铜片(要去除铜片表面的杂质和氧化物,用实验水冲洗除酸,并用丙酮清洗,再用氮气吹干待用,临用前处理,保持表面光亮)进行除硫,防止干扰目标物。多环芳烃大部分是无色或淡黄色的结晶,个别具有深色。沉积物经过[/font]48[font=宋体]小时的索氏提取,当提取液颜色呈现无色,有可能是样品的基体相对简单,杂质少,也可能是多环芳烃的含量比较低或含有无色的多环芳烃物质;当发现有黄色的情况时,也加了足量的铜片,则一种可能是基体复杂,有色素等的杂质存在,一种可能是多环芳烃含量比较高引起颜色的加深。如果是杂质多的话,可通过净化步骤进行消除干扰。[/font]以上内容来自仪器信息网《样品前处理实战宝典》
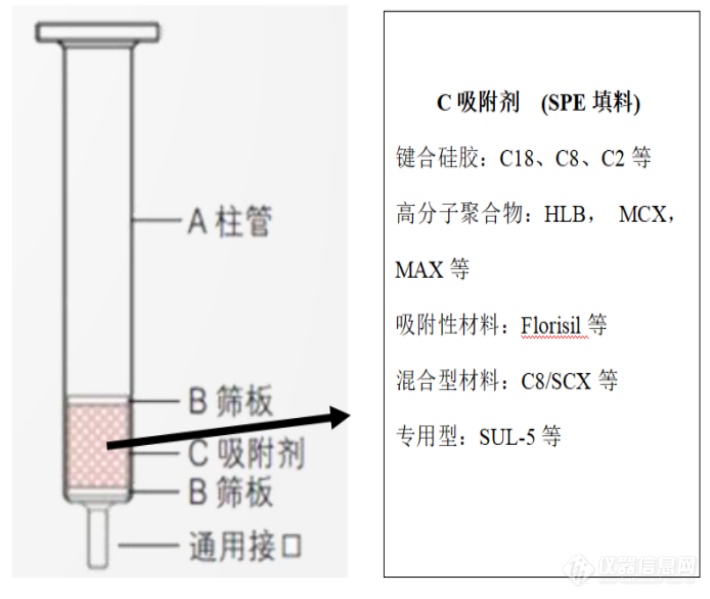
[b] 以多环芳烃为船,来固相萃取的世界遨游一番前言[/b]固相萃取(Solid Phase Extraction, SPE)是一种基于色谱分离的样品前处理方法。越来越多地应用在环境、医药、化工等领域。我们先看一下固相萃取柱的结构示意图。[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152046051298_8342_3247983_3.png!w690x582.jpg[/img]特点:A柱管有1ml, 6ml等不同大小的规格,C吸附剂是固相萃取核心,不同的目标化合物,用途不同,填料种类不同,质量也不同,比较常见的有100mg,500mg和1g。颗粒大小为40-50μm,填料规格是不规则的,与液相色谱相比分离效率低。多环芳烃是环境领域重要的污染物,水质中多环芳烃的萃取,土壤中多环芳烃的净化都要用到固相萃取。[b]1、反相色谱柱特征和原理[/b]水质中多环芳烃萃取用的是反相固相萃取柱,多用C18柱,它的特点是固定相极性流动相极性(水溶液)。[img=,690,633]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152048268798_241_3247983_3.png!w690x633.jpg[/img]从微观图看出,填料呈不规则排布,原子结构,无定型的多孔基质,硅原子与氧原子相结合形成“硅氧键”即(Si - O - Si)[color=#323232][b]2、[/b][/color][b]什么是固相萃取柱封端?封端的意义是什么?[/b]以下是封端示意图[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152049320138_8208_3247983_3.png!w690x582.jpg[/img]硅胶表面的硅羟基(Si-OH) 由于空间位阻的存在,硅羟基与化合物的极性基团存在极性相互作用和离子交换作用,在化合物的保留时增加了其它不需要的作用力,用短链氯硅烷(如三甲基氯硅烷)键合活性的硅羟基,种操作被称为封端。封端:简单说就是消除不需要的极性基团间的作用力,在萃取时得到更好的效果。[b]3、水质中多环芳烃固相萃取两个重要步骤[/b]3.1大体积进样,多环芳烃中的碳氢键同硅胶表面的官能团,非极性相互作用,吸附在Si - O - Si键上,因此多环芳烃保留在CI8柱上。保留示意图如下:[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152051164078_1766_3247983_3.png!w690x582.jpg[/img]3.2洗脱,用极性溶剂将多环芳烃从Si - O - Si键上分离开,多环芳烃的洗脱一般选用二氯甲烷用为洗脱剂,它的极性指数是3.4。它的吸附作用大于吸附剂与多环芳烃的作用力(范德华力)。有同学可能会问,为什么不用极性更强的如丙酮(极性指数5.4)、甲醇(极性指数6.6)进行洗脱,根据相似相溶原理,二氯甲烷对16种多环芳烃溶解性更好,特别是高环的多环芳烃。[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152052403598_4124_3247983_3.png!w690x582.jpg[/img][b]4、反相固相萃取注意事项[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907160922441125_4546_3247983_3.png!w690x582.jpg[/img][img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152054197588_8894_3247983_3.png!w690x582.jpg[/img][img=,690,538]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152058168985_7176_3247983_3.png!w690x538.jpg[/img]5、正相色谱柱特征和原理[/b]5.1土壤中多环芳烃净化用的是正相固相萃取柱,以吸附型填料Florisil为代表[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152059060415_5326_3247983_3.png!w690x582.jpg[/img][color=#333333]结构示意图中,石墨中的碳原子呈六元环状的平面层状结构,这样的结构优点是:吸附非极性和弱极性的化合物又可以吸附极性化合物,对化合物表现出很广的吸附谱。[/color]用在土壤净化中,土壤中的杂质以腐质物质为主,它的作用是吸附大量的腐质物质等,达到去除基质(杂质)干扰的效果。[img=,690,560]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152059526188_4940_3247983_3.png!w690x560.jpg[/img]土壤样品用正相固相萃取柱净化后,一是背景值低,定性和定量更准确,二是减少对仪器的污染,特别是进样口和离子源([url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]分析时)。5.2净化步骤主要分为三步。[img=,690,582]https://ng1.17img.cn/bbsfiles/images/2019/07/201907152100391005_7636_3247983_3.png!w690x582.jpg[/img]活化时与反相固相萃取柱同样溶剂不能流干,否则易形成沟流,降低除杂能力。5.3正相固相萃取与反相固相萃取区别与反相固相萃取不同的是,萃取柱上留住的是杂质,收集多环芳烃的淋洗液。上样的体积控制在2-5ml左右,上样体积过大,杂质容易被有机溶剂冲下来。淋洗时,用V正己烷(极性指数0.06):V丙酮(极性指数5.4)=9:1(体积比),通过公式P’=ΦaPa+ ΦbPb可以算出混合溶剂极性指数约为0.6,既可以把多环芳烃淋洗下来,又不至于极性过强,把杂质大量带下来。[b]小结:[/b]1、固相萃取是用途广泛的萃取方法,在建立新方法时,首先要考虑基底特性,是水、油,还是其它介质,查询相关的文献可以获得。2、判断分离机制,是极性还是非极性。即选择对目标有明显较强保留的SPE柱。3、淋洗时,考虑淋洗液的极性,可以通过查表(溶剂极性表),混合溶剂通过公式计算出它的极性,获得较为满意的淋洗和洗脱效果。 不妥之处,望各位老师批评指正!
想做肉制品中除了国标法规定的16种多环芳烃之外的其他种类的非定向筛查,大概就是提取净化后全扫看看有没有新物质,有新物质就把新物质通过某种方法(目前考虑半制备)分离成单一组分后核磁红外测结构,推断结构式。请问有大神做过么?目前查文献没人做过这个思路的。或者相关领域有所涉猎的,请各位大神不吝赐教
各位大神好,目前我在做食用油中多环芳烃检测,由于实验室仪器限制只能用HPLC-UV和GC-FID检测,之前没做过这个所以还是一头雾水现在有的仪器有C18 和MgSiO4的SPE柱子。 我原本是按GB/T动植物油脂中多环芳烃检测的前处理方法试了下处理我的油样(炸东西炸了一段时间的锅里残留油)先液液萃取然后两次SPE净化吹到50μL左右加乙腈稀释到250μL。然后去跑HPLC, 参数流速是2.0ml/min\进样20μL\波长254nm\梯度乙腈60% 水40% 最后乙腈100%,跑40分钟,出来的峰看得我是云里雾里。请问我怎么改进萃取净化方案比较好?我看有些文献上是直接不液液萃取直接上SPE吹干这样,这样杂质能除得干净吗?GC还没做,所以这里先不讨论……
求…固体废物多环芳烃原子荧光谱图,做对比~
做的是多环芳烃EPA16,目前有[font=&]氘代内标(菲-D10,芘-D12),想用菲-D10(3环)来校正2-3环的多环芳烃(在开始时加入,每次计算回收率校正数据),用芘-D12(4环)来校正4-6环多环芳烃,请问这种做法可以吗?各种氘代多环芳烃内标具体可以监测那些多环芳烃呢?有无明确要求呢?是每一个环数的多环芳烃都要有氘代内标吗?(高环数氘代内标太贵了)[/font]
敢问各位高手,我用液相做多环芳烃(16种)时,芴和菲怎么都分不开,谁能指教一下?(流动相为乙腈和水的梯度洗脱)
我最近在做关于某种细菌对多环芳烃降解率的摇瓶实验,只用了菲、芴、芘、荧蒽四种多环芳烃,在测溶液中多环芳烃残留量的时候,还需要加替代和内标吗?加的种类和测16种多环芳烃时一样吗?我们课题组以前都是做土壤重金属的,师兄师姐也不知道,文献里也没有说明白,很迷茫测16种多环芳烃时我用的替代是2-氟联苯和对三联苯混合液,内标是苊、菲、屈、苝的氘代物
最近一直在做鞋子主要是鞋底材料的多环芳烃的检测,我们是用正己烷来提取,然后过滤,旋转蒸发,氮吹至干,甲醇定容,HPLC检测分析,可是出来的峰好乱,杂质多,跟标准不好对照定性啊,请大家指教预处理过程啊,急待回音.......这几天用索氏提取,震荡提取,都没什么效果,不知道该怎么净化啊
本人最近刚接触到多环芳烃,所以有很多要与各位高手交流一下。首先是标准溶液的配制问题:因为多环芳烃的溶解度普遍较小,我主要做了:萘、菲、荧蒽和蒽几种多环芳烃,萘和菲还能用紫外检测出来,只是菲的吸光值较小(在其溶解度一下浓度的吸光值在0.3以下),而荧蒽和蒽几乎检测不出来。有人建议我用荧光检测,但对其不了解,因为我们实验室没有荧光光度计。希望各位高手能给我一点意见,或是对多环芳烃的定量分析有什么好的建议也行!十分感谢!
近期领导下任务独立完成GC面积归一化法测对二甲苯纯度及杂质含量的不确定度评估,因为接触的少,根本没有思路下笔,各位大神没有做过类似的评估或者能够分享一下相关资料或文章可以借鉴一下对二甲苯:PX主要杂质有:非芳烃,苯,甲苯,乙苯,间二甲苯、正丙苯、邻二甲苯、异丙苯、C9、C10、PDEB望各位大神能帮小弟一把
多环芳烃用气质联用分析时,内标物选什么?
请问用GC-MS测堆肥中的多环芳烃时应该怎么对堆肥做预处理?急用,谢谢
有没有老师用过岛津的RF-10AXL型号的荧光检测器做水中的多环芳烃,自己走了很多针都只有14个峰,想参考参考做过这个方法的流动相和检测器的梯度是咋个设置的
本人在尝试建立水样中多环芳烃的GC-MS测定方法,参考的方法是国标HJ 646-2013《环境空气和废气 气相和颗粒物中多环芳烃的测定 气相色谱-质谱法》,其中遇到两个问题不是很清楚,特向各位前辈求助:1. 方法中采用2-氟联苯和对三联苯-d14作为替代物,在样品前处理之前加入已知的量,再利用前处理后样品的测定值计算出提取回收率,从而可以评价前处理过程产生的损失。那么,多环芳烃的测定值是否也要用这个提取回收率反算出原有值?还是直接用测定值作为最终结果呢?2. 方法要求制作硅胶层析柱进行样品的净化,请问用二氯甲烷/正己烷洗脱后的柱子还能重复用于净化另一个样品吗?还是要除掉填料重做层析柱?如果可以重复用,是不是也要进行一定的处理?用二氯甲烷或正己烷冲洗可以吗?可能这些问题在各位看来比较初级,不过作为初次接触此行的小弟来说还是比较头疼的,烦请您指点一二!谢谢~
[font=微软雅黑]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-4153.html[/url]多环芳香烃(PolycyclicAromatic Hydrocarbons,简称PAH或PAHs)又称多环性芳香化合物或多环芳香族碳氢化合物,是芳香族碳氢化合物的一种特例,由不包含杂环或取代基的芳香环所组成;其致癌、光致毒、破坏免疫系统等对人体造成巨大危害。[/font][font=微软雅黑]首先,多环芳烃最明显的危害来至于其致癌性,多环芳烃的致癌性可能是因其易于和细胞内的DNA、RNA等遗传物质结合而起致癌作用。[/font][font=微软雅黑]其次,多环芳烃更危险在于它们暴露于太阳光中紫外线辐射时的光致毒效应。太阳光中可见光区和紫外光区的光对多环芳烃的毒性有显著影响。[/font][font=微软雅黑]最后,多环芳烃可以引起机体的免疫抑制反应,表现为血清免疫学指标的改变。[/font][font=微软雅黑]鉴科检测参考《HJ 784-2016 土壤和沉积物多环芳烃的测定高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]》,建立了利用全自动固相萃取仪(Fotector Plus)结合高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]检测沉积物中多环芳烃残留量的方法。在100mL丙酮-正己烷(1+1)提取后,使用Auto EVA-08IR浓缩至1mL后 Fotector Plus全自动固相萃取仪净化,自动完成 SPE 柱活化、样品上样、淋洗、收集等步骤,收集液再氮吹浓缩、溶剂转换、定容后,用HPLC检测。[/font][font=微软雅黑][/font]
试验室进行了橡胶中多环芳烃(蒽,荧蒽,芘,菲)能力验证实验,但是一直没有得到目标结果,前来向各位请教,将细节详细列出,(如不全,后续补充)希望得到各位指点!收到的样品是橡胶粉末,可直接使用。实验参考AfPS GS 2014:01 PAK.前处理:称取0.5g样品包在称量纸里,放在20ml小瓶中加入10ml乙酸乙酯或甲苯,拧紧瓶盖于60℃下超声萃取1h,然后恢复室温后萃取液全部移出稀释,大约稀释到500-1000倍,使其落在0.1-1ug/ml浓度范围内。然后[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]进样分析,使用MS定量。标准曲线:多环芳烃标液是购买的有证的混合标液,浓度为1000ug/ml,溶剂为二氯甲烷。使用自动配标仪进行样品稀释,得到0.1-1ug/ml浓度的标液(0.1,0.3,0.5,0.7,1.0),得到的标准曲线效果并不是很好,线性基本在99.8-99.9%,少有超过99.9的。有时某个点偏离,刨除之后4个点能超过3个9.结果:橡胶中多环芳烃含量在100-800mg/kg,我们得到的结果偏大居多,有时能大百分之二三十。分析:1 我们得到的结果不稳定,时高时低,有时前两种得到了符合要求的结果,下次做另两种又得到符合结果而这两种又不符合了。所以这表明萃取效果不稳定,并不是每次都能得到稳定量的四种物质。所以可能是什么因素影响萃取效果呢?2 标准曲线对结果影响很大,所以我们得到的标准曲线可以用吗?包括线性没超过999的以及四个点的曲线。为什么得到的标曲线性并没有很好呢?对于二氯甲烷这种易挥发的溶剂,我们应该如何操作得到符合要求的标准曲线呢?还有打开的母液放在2ml进样瓶中拧紧封好封口膜放在4度冰箱里几天之后还可以用吗?3 MS定量时菲和蒽靠的比较近,基线不平,是否需要修正基线为曲线的起点至终点?如下图感谢大家的指点,目前问题基本得到解决,简单介绍一下处理方法。首先我们的样品中多环芳烃含量很高,而且基于样品基质均匀性的考虑,取0.5g样品超声萃取。萃取得到的多环芳烃总量比较大,避免多次稀释带来的误差,仅稀释到100ml。因为之前参考标准使用的是不分流选择扫,这适合于含量小的物质,而不适合我们的样品,因此调高进样口温度,使用分流进样以及全扫模式,这样可以适用于浓度相对较大的样品,最后可以得出比较稳定的效果,四种物质有三种均落在误差区间内,仅有菲仍小于给定值,目前仍在解决。多同各位前辈,仪器工程师交流请教,的确可以给我们带来解决思路,再次感谢大家!
实验室新开发项目要做多环芳烃的测试,现在要我们自行研究多环芳烃测试,前处理做法以及其标液的配制,有哪位同仁已经做过或在做多环芳烃测试,就帮助。
分析人体组织中的多环芳烃和邻苯二甲酸酯时,相对于样品中的浓度,空白值从百分之十几到百分之一百的都有;后来经过计算,按照北京市大气中气态多环芳烃100ng/m3计,十几升空气中的多环芳烃即相当于样品中的空白值。请教可以如何消除实验室本底值?建立一个具有空气过滤系统的超净实验室似乎不太现实;可以使用手套箱或者超净实验台一类的装置吗?但这这样有一个问题,因为过程中要进行旋转蒸发浓缩和过几十厘米高的层析柱等操作,不知能否把这些仪器都放进去。在美国做实验,尽管多溴联苯醚有空白,但终究比样品低一两个量级;现在多环芳烃的本底值已经高得不能忍了……欢迎大家讨论~~谢谢~~~
求教:如题,大家做雾霾中16种多环芳烃时防护和注意事项。1,标准溶液稀释过后的使用液如何保存?需要特殊的瓶子吗,没见过操作上讲的带聚四氟乙烯沉淀的螺纹玻璃瓶。有人能提供一下图片吗。2,操作过程废弃物如何处理,扔到哪里,3,这16种多环芳烃,致癌性极大,到底该如何防护,有没有比较好的建议,咱们接触的量到底有多危险。
脂肪酸甲脂、多环芳烃英文怎么写?谢了!







 我要推广仪器
我要推广仪器
 下载APP
下载APP