介绍了XPS表征纤维样品的三种制备方法,并通过这三种制备方法对三类不同尺寸的纤维样品进行XPS分析及结果对比,进而展示三种制备方法的特点及适用范围。
纳米材料的概述、制备及其结构表征是一个小型的综述[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=63065]纳米材料的概述、制备及其结构表征[/url]
纳米氧化铋的制备及表征,10积分该论文有吗
[font=&]【题名】:单价阳离子选择性分离膜的制备与表征[/font][font=&]【全文链接】: https://cdmd.cnki.com.cn/Article/CDMD-10358-1014432246.htm[/font]
蛋白质的翻译后修饰常常会影响蛋白质的结构和功能,反映在生物制药工业上,会对药品的安全性和有效性产生重大影响。翻译后修饰常常表现为电荷变异体,因此电荷异构体的分析成为了质量控制的一个关键项。目前常见的电荷异构体分析方法为IEF/cIEF或iCIEF,可以鉴别生物药,对生物药的纯度进行分析,测定电荷异构体的等电点以及各种异构体的分布。但是,等电聚焦或者毛细管等电聚焦存在很多短板,最明显的就是无法大规模制备异构体。美国基因泰克公司的科学家曾经用一种叫做自由流电泳的工具,高分辨率高通量大规模对单抗的电荷异构体进行分离制备,并结合各种分析手段,对每一个异构体进行了深度表征。现分享论文如下,欢迎大家讨论!
作者:贾文娟题目:药物缓释微球的制备、表征及性能研究期刊:四川大学年份:2007链接:http://www.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&QueryID=4&CurRec=37&dbname=CMFD9908&filename=2008027766.nh&urlid=&yx=
作者:何冰题目:5-氟尿嘧啶壳聚糖微球的制备与表征期刊:天津大学年份:2007链接:http://www.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&QueryID=4&CurRec=186&dbname=CMFD0911&filename=2008186937.nh&urlid=&yx=
【序号】:2【作者】:祁凤英1,2王蕾2李东东2【题名】:可缓释富血小板血浆生长因子的新型自组装多肽水凝胶制备及性能表征【期刊】:中国组织工程研究 . 【年、卷、期、起止页码】:2024 ,28 (15) 【全文链接】:
【序号】:1【作者】: 郝建文1戴晨伟1刘永春【题名】:磺化CS/PVA交联阳离子交换膜的制备与表征【期刊】:塑料. 【年、卷、期、起止页码】:2018,47(01)【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C44YLTlOAiTRKibYlV5Vjs7i0-kJR0HYBJ80QN9L51zrP58BGoMgeIi71m5UMIOqJHEwFDVRvjtk0J6UYaAFTvtk&uniplatform=NZKPT
【序号】:4【作者】: 程鸿昊【题名】:新型水溶性壳聚糖的制备、结构表征及性能分析【期刊】:深圳大学【年、卷、期、起止页码】:2015【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201502&filename=1015419049.nh&uniplatform=NZKPT&v=QgCQWC8E7NV0DuUWCarIReSDOH9hlLxapcjlUYLQPTNi7WCZwmL_ofmQU5cEmZ-9
【序号】:3【作者】:潘晴彦1,2周闯2杨子明2【题名】:季铵盐修饰壳聚糖及其复合膜的制备与表征【期刊】:现代食品科技. 【年、卷、期、起止页码】:2021,37(12)【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C44YLTlOAiTRKibYlV5Vjs7iJTKGjg9uTdeTsOI_ra5_Xd00vMdAPh0TCP4aZG7LDh5g0EkC7XCgPTl5eG7LeY5W&uniplatform=NZKPT

氧化石墨烯改性双极膜中间层的制备与表征摘要 环境污染也成为了人类目前最大的问题,在许多化工企业中,所排放的废水中会含有较多的盐,传统工艺过程对这些废水的处理(萃取、中和、吸附和生化等)存在缺陷,工艺过程费用高,但是这种处理方法的成本费用很高,但是引用双极膜电渗析技术,这种处理废水的工艺技术不但能够节省费用且工艺过程也更简单。双极膜电渗析技术可以从极稀醋酸废水中提取出醋酸,与传统的电渗析技术相比,回收的浓缩液可以达到36%以上,而传统的浓缩到20%,超过20%以后,醋酸的浓度降低到0.1%时候,电压就会急剧上升。1. 引言 双极膜技术起源于50年代,但是那时候双极膜技术不成熟,发展缓慢,膜性能非常差,经过60多年的发展研究,双极膜技术得到了快速发展,随着对双极膜水解机理认识逐渐加深,双极膜的制备也从简单的层压型、单片型转变到界面层复杂结构,双极膜的跨膜电压得到很大的降低,80年代中期,通过美国科研人员的努力,双极膜的中间层得到非常有效的改进,随后国内外双极膜研究工作者继续对双极膜中间层进行改进,提出了多种中间层催化水解物质(PEG、PVA等),使得双极膜的性能得到优化,电阻也大幅度降低,膜性能、机械强度、能耗方面有了较大的改进,技术应用也从化工污染治理、资源回收、食品工程、能源、环境、生命科学扩展到多个领域,为许多领域的技术难题带来了新的工具。目前,国外对双极膜的研究较为深入,双极膜技术在美国、日本应用已经非常广泛,且开始商品化,国内目前还处于研究时期,许多问题需要解决和突破。http://ng1.17img.cn/bbsfiles/images/2015/09/201509211532_566971_2984502_3.jpg2. 膜材料、实验试剂与仪器2.1 膜材料 本实验采用的阴离子交换膜在江千秋环保水处理有限公司购置,其材料参数见表1.http://ng1.17img.cn/bbsfiles/images/2015/09/201509211534_566972_2984502_3.jpg2.2 实验试剂 本实验所用试剂见表2。http://ng1.17img.cn/bbsfiles/images/2015/09/201509211536_566973_2984502_3.jpg2.3 实验仪器 本实验所用仪器见表3。http://ng1.17img.cn/bbsfiles/images/2015/09/201509211537_566974_2984502_3.jpg3.实验过程3.1 双极膜的制备 PAAS/GO/AM-1双极膜是以商业化的阴离子交换膜AM-1作为双极膜的基膜层,并在其表面喷涂氧化石墨烯,形成中间层,然后再喷涂戊二醛和聚丙烯酸钠交替喷涂形成阳离子交换层。制备双极膜的流程如图2-2所示,实验步骤如下所示:(1)将买来的阴离子交换膜AM-1膜剪成10×10cm2的方块,放在1mol·L-1NaCL溶液中24小时;(2)取出浸泡的阴离子交换膜,然后将其移到1mol·L-1 NaOH的溶液中放置6 h,过后再用用蒸馏水洗净,然后再将上述膜放到1mol·L-1 HCl浸泡6 h,使用去离子水清洗膜表面,而后将清洗后的膜放置到40℃的恒温烘箱中烘干;(3)将阴离子交换膜(AM-1)固定放置到载膜器上,开始调节载膜器的电机到恒定转速;(4)调节压力至 0.40MPa 下,将配置好的氧化石墨烯溶液喷涂到阴离子交换膜上,喷涂一定的次数形成界面层,再交替喷涂戊二醛(GA)和聚丙烯酸钠(PAAS),通过喷涂戊二醛和聚丙烯酸钠一定的次数形成阳离子交换膜; 喷涂结束以后,将双极膜取下,放置到干净的恒温烘箱内,干燥一段时间后即可得到PAAS/GO/AM-1双极膜。http://ng1.17img.cn/bbsfiles/images/2015/09/201509211539_566976_2984502_3.jpg3.2 双极膜的性能表征 双极膜I-V主要用来表征膜电流-电压的基本特性,可以通过I-V曲线反映出双极膜的能耗大小、水解离速率、水解离电压和极限电流密度等。本论文中I-V测试装置如图2-3所示,通过使用Nafion膜使阴、阳极室与双极膜分开,这样有效的阻挡了电极产物与PAAS/GO/AM-1双极膜之间的接触。双极膜放置在测量池中,通过调节水泵,控制盐溶液在在测量池中恒速循环,测量体系所用的溶液均为0.5mol·L-1Na2SO4溶液。双极膜的电压通过使用两Ag/AgCl电极和万能表测量得出。http://ng1.17img.cn/bbsfiles/images/2015/09/201509211542_566981_2984502_3.jpg
【序号】:1【作者】:孙陆军【题名】:磷酸钙/明胶多孔支架的制备与表征【期刊】:【年、卷、期、起止页码】: 北京化工大学, 高分子化学及物理, 2008, 硕士【全文链接】:http://www.cnki.net/KCMS/detail/detail.aspx?QueryID=1&CurRec=43&recid=&filename=1014450337.nh&dbname=CMFD201501&dbcode=CMFD&pr=&urlid=&yx=&v=Mjc0NDZZUzdEaDFUM3FUcldNMUZyQ1VSTHlmWnVScUZpRG1WcnJJVkYyNkdyZTlIdExQcUpFYlBJUjhlWDFMdXg=

[align=left][font='times new roman'][size=16px]SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-DMOA[/size][/font][font='times new roman'][size=16px]色谱固定相的制备[/size][/font][font='times new roman'][size=16px]及表征[/size][/font][/align]SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相的制备分为环氧基修饰二氧化硅的制备和十八叔胺修饰二氧化硅的制备。球形多孔硅胶在100℃下真空干燥24 h以除去硅球表面吸附的水分,备用。第一步为环氧基化二氧化硅(SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS)的制备:通过硅烷偶联反应制备环氧基二氧化硅。首先称取2.4 g干燥过夜的球形硅球置于250 mL圆底烧瓶中,依次加入30 mL无水甲苯、2.4 mL GPTMS和300 μL三乙胺,N[font='times new roman'][sub][size=16px]2[/size][/sub][/font]氛围下95℃下加热回流24 h。反应结束后,依次用甲苯、乙腈和甲醇洗涤两遍以洗去杂质,抽提。置于80℃烘箱中干燥过夜,获得SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS。第二步为十八叔胺修饰二氧化硅色谱固定相的制备:称取2.4 g SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS置于250 mL的圆底烧瓶底部,加甲醇30 mL重悬,随后加入12 mL的DMOA,充分混匀后充入氮气,50℃下加热回流2 h。反应结束后,用大量甲醇洗去反应体系中的杂质,置于80℃烘箱中,烘干24 h,获得SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相。[align=center][size=13px]图[/size][size=13px] SiO[/size][font='times new roman'][sub][size=13px]2[/size][/sub][/font][size=13px]-DMOA[/size][size=13px]色谱固定相制备示意图[/size][/align][align=left][font='times new roman'][size=16px] SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-DMOA[/size][/font][font='times new roman'][size=16px]色谱柱的填充[/size][/font][/align]采用高压匀浆法填充SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱柱。称取2.0 g的SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相置于100 mL烧杯中,将SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱填料与匀浆液混合,超声处理两秒使其均匀分散,匀浆液按照氯仿-环己醇(3:7,v/v)比例进行配置。将混悬液倒入空柱管(150 mm × 4.6 mm)的匀浆罐中,连接好装置避免漏液。调节压力上升至35 MPa,加压液甲醇-异丙醇(1:1,v/v)会随着管路流过匀浆罐下方的色谱柱,在此压力下维持30 min,降低压力为0 MPa,静置30 min。将柱管卸下后装入筛板和柱头后,用乙腈冲洗色谱柱,备用。[align=left][font='times new roman'][size=16px]SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-DMOA[/size][/font][font='times new roman'][size=16px]色谱固定相表征[/size][/font][/align]首先,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相采用傅里叶变换红外光谱仪对其进行表征。图a为SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA固定相在4000-400 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]范围内的红外光谱图。图a中可以明显看到在SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA的红外谱图中1100 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]处的强吸收峰,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA是以二氧化硅为基底,所以此处峰可以认定为Si-O的伸缩振动所引起的。在SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS谱图中,由于980 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]处峰的消失,可以认定为硅球中的硅醇基团都被GPTMS掩盖,证明GPTMS已经成功键合到SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]上。相比于SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS,可以在SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA固定相中观察到2930 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]和2850 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]两处吸收峰,这属于DMOA结构中典型的甲基和亚甲基的特征峰。此外,对SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相进行热重分析。如图3-2b所示,在温度上升至800℃,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]几乎没有质量损失。SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS在400℃质量损失加剧,可以归于SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS表面键合硅烷化试剂损失导致。SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相大约在170℃开始第一次发生质量损失,这可能是SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相表面最外层键合的DMOA损失导致的,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相在400℃开始第二次质量损失,这与SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS的质量损失相似。在800℃时,与SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]相比,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-GPTMS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA质量分别损失了13.5%和17.0%。上述结果可证明SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱固定相已成功制备。 [img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211162158483591_7554_5389809_3.png[/img][align=center][size=13px]图[/size][size=13px]硅球、[/size][size=13px]SiO[/size][font='times new roman'][sub][size=13px]2[/size][/sub][/font][size=13px]-GPTMS[/size][size=13px]和[/size][size=13px]SiO[/size][font='times new roman'][sub][size=13px]2[/size][/sub][/font][size=13px]-DMOA[/size][size=13px]的红外光谱图([/size][size=13px]a[/size][size=13px])和热重分析图([/size][size=13px]b[/size][size=13px])[/size][/align][align=left][font='times new roman'][size=16px] SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-DMOA[/size][/font][font='times new roman'][size=16px]色谱柱保留机制[/size][/font][/align]首先选取苯、乙苯、丙苯、丁苯对SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱柱的保留机制进行考察。从图可以看出,当洗脱液中ACN含量从40%增加到90%时,四种烷基苯类物质的保留呈下降趋势,四种化合物的保留因子从7.83下降到0.05,表现出典型的反相色谱保留机制。由于SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA固定相表面存在季胺基团,因此SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱柱可同时提供亲水保留机制。进一步选择三种亲水性不同的酰胺类物质(2-碘乙酰胺、对氨基苯甲酰胺和烟酰胺)考察其保留机制。如图3-3b所示,随着洗脱液中H[font='times new roman'][sub][size=16px]2[/size][/sub][/font]O含量从1%增加到40%,2-碘乙酰胺、对氨基苯甲酰胺和烟酰胺的保留因子逐渐下降,表现出典型的亲水色谱保留机制。因此,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-DMOA色谱柱具有反相/亲水混合模式保留机制。[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211162158486956_7166_5389809_3.png[/img]
反相色谱制备分离后,如何快速从流动相中得到产品?
反相色谱制备分离后,如何快速从流动相中得到产品?
反相色谱制备分离后,如何快速从流动相中得到产品?
反相色谱制备分离后,如何快速从流动相中得到产品?
反相色谱制备分离后,如何快速从流动相中得到产品?
反相色谱制备分离后,如何快速从流动相中得到产品?
【序号】:3【作者】:祁凤英1,2王蕾2李东东【题名】:可缓释富血小板血浆生长因子的新型自组装多肽水凝胶制备及性能表征【期刊】:中国组织工程研究. 【年、卷、期、起止页码】:2024,28(15)【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=6xaVI2TORM1swCWGB30yfUSs7gagqZ8OuIiWkV2_s1SHFu-K1KszuTVN3jiF4Ab9hQrTPCOoiwTAoBUscEZgIGyLDzwVbBMulNObHUFG5Ayp20EWIPttGstjxBJ37fe743uLcw4DVrM=&uniplatform=NZKPT&language=CHS
【序号】:8【作者】: 陈欣1卢小菊1,2孟鸳1【题名】:聚乙烯吡咯烷酮/壳聚糖水凝胶的制备与表征【期刊】:湖北理工学院学报. 【年、卷、期、起止页码】:2016,32(04)【全文链接】:[url]https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2016&filename=HSGD201604007&uniplatform=NZKPT&v=Ur3uQPHp3CF41AAdzeLXFbOBIluj4tRQZqNKITVAm6QqBA6_xZS8rXbC1tFdRSdn[/url]
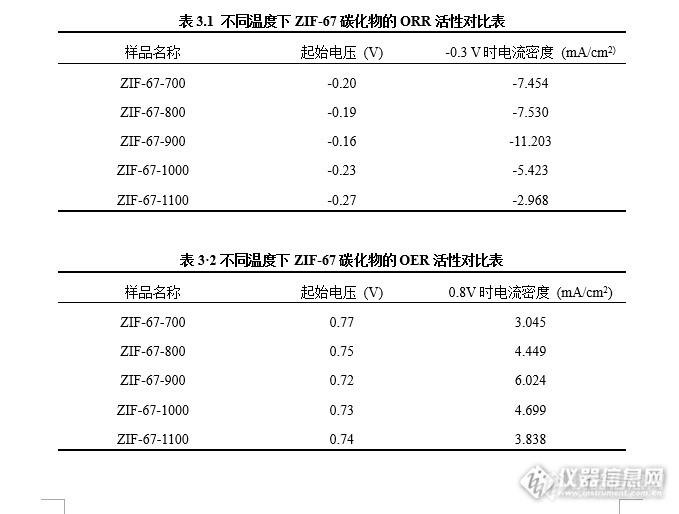
不同温度下ZIF-67碳化物制备及表征1.1基于ZIF-67的碳化物制备 将ZIF-67,研磨至微小粉末状;称取一定量的ZIF-67装入小瓷盅内,在通入N2的条件下,在高温炉内以10 ℃/min的速率升温至一定温度,并维持5 h,自然冷却后取出,研磨。 本实验将ZIF-67分别在700、800、900、1000、1100 ℃温度下进行直接碳化,共制得五个样品。下文用(ZIF-67-700,ZIF-67-800,ZIF-67-900,ZIF-67-1000,ZIF-67-1100表示)图1.1是基于ZIF-67的碳化物制备流程图:http://ng1.17img.cn/bbsfiles/images/2016/08/201608291032_607212_2984502_3.png2结构表征2.1 XRD 图2.1为ZIF-67分别在700、800、900、1000、1100℃下的碳化物的XRD谱图对比图。如图所示,不同温度下进行碳化,所得ZIF-67碳化物的晶型结构基本一致。在44°、51 °、76 °附近,所有温度下的ZIF-67碳化物都有三个较为明显的峰,通过与Co的XRD谱图进行对比,峰值的强度和出现位置基本相吻合,可能含有金属Co;另外,在26°附近,也存在明显的峰,通过与C的XRD谱图进行对比,峰值的强度和出现位置基本相吻合,可能含有碳。所以得到的产物可能是负载Co的纳米多孔碳,但还需要进一部验证。http://ng1.17img.cn/bbsfiles/images/2016/08/201608291033_607213_2984502_3.png3电化学测试3.1 电极的制备与工作环境 本实验选择Ag/AgCl电极作为参比电极 (武汉高仕睿联有限公司),其电势相对于标准氢电极 (NHE)为0.197V。选择碳棒为辅助电极。采用负载催化剂的东丽20%疏水性碳纸为工作电极和负载催化剂的玻璃电极头为工作电极。选择电解池为三电极体系电解池。 碳纸工作电极的制备:首先将碳纸裁成2×1 cm2大小,并用无水乙醇冲洗。取2.5 mg待测样品,加入2.5mg Vulcan XC-72活性炭,分散于1 mL无水乙醇中,并加入50 μL Nafion溶液,超声搅拌至均匀分散后,形成工作电极的活性材料。使用移液枪取100 μL活性材料均匀铺于碳纸1×1cm2内。50 ℃下干燥30分钟,得到工作电极。RDE工作电极的制备:首先使用金相砂纸,粒度为0.3 μm的Al2O3抛光粉将玻璃电极打磨抛光,并用无水乙醇冲洗干净。取2.5 mg待测催化剂样品,再加入 2.5mg Aulcn XC-72活性炭,分散于1 mL无水乙醇中,并加入50 μL Nafion溶液,超声搅拌至均匀分散,形成工作电极的活性材料。使用移液枪取21 μL活性材料均匀铺于面积为0.196cm2的玻璃电极头上,50 ℃下干燥30分钟,得到工作电极。碳纸测试环境:在0.1 M KOH为电解液中,使用电化学工作站进行碳纸测试。(1) 首先在N2饱和环境下,分别以50 mV/s、5mV/s的扫描速率进行背景测试,然后在O2饱和的环境下以5 mV/s的扫描速率进行ORR测试。(2)在N2饱和环境下以5 mV/s的扫描速率进行OER测试。RDE测试环境:在0.1 M KOH为电解液中,使用电化学工作站进行旋转圆盘电极测试。(1) 首先在N2饱和环境下,分别以50 mV/s、5mV/s的扫描速率进行背景测试,然后在O2饱和的环境下以5 mV/s的扫描速率在2025rpm,1600 rpm,1225 rpm,900rpm,625 rpm,400 rpm的转速下分别进行ORR测试。(2)在N2饱和环境下以5 mV/s的扫描速率在1600rpm的转速下进行OER测试。3.2不同温度下ZIF-67碳化物的催化活性比较 不同温度下ZIF-67碳化物的ORR与OER催化性能分别由图3.1和图3.2表示。 如图3·1和表3·1所示,在ORR中,ZIF-67-700碳化物的起始电压为-0.20V,-0.3 V时电流密度是-7.454 mA/cm2,ZIF-67-800碳化物的起始电压为-0.19V,-0.3 V时电流密度是-7.53 mA/cm2,ZIF-67-900的起始电压为-0.16V,-0.3 V时电流密度是-11.203 mA/cm2, ZIF-67-1000碳化物的起始电压为-0.23V,-0.3 V时电流密度是-5.423 mA/cm2, ZIF-67-1100碳化物的起始电压为-0.27V,-0.3 V时电流密度是-2.968 mA/cm2。相比之下,ZIF-67-900起始电压低出很多,-0.3V时电流密度也更大,拥有最好的催化性能。 如图3.1和表3.1所示,在ORR中,ZIF-67-700的起始电压为0.77V,0.8 V时电流密度是3.045 mA/cm2,ZIF-67-800的起始电压为0.75V,0.8 V时电流密度是4.449 mA/cm2,ZIF-67-900的起始电压为0.72V,0.8 V时电流密度是6.024 mA/cm2,ZIF-67-1000的起始电压为0.73V,0.8 V时电流密度是4.699 mA/cm2,ZIF-67-1100碳化物的起始电压为0.74V,0.8 V时电流密度是3.838 mA/cm2。相比之下,ZIF-67-900,拥有最低的起始电压,0.8V时电流密度也最大,有最好的OER催化活性。 上述结果过说明,不同温度下碳化ZIF-67,900 ℃下碳化得到的ZIF-67碳化物拥有更好的催化性能,相比其他温度下的碳化物,ZIF-67-900起始电压较小,电流密度也最大,在ORR和OER中都展现了良好的催化性能,说明了900 ℃可能是利用碳化过程优化ZIF-67催化剂的最适宜温度,也再一次证明了ZIF-67碳化物是性能优良的双功能催化剂。http://ng1.17img.cn/bbsfiles/images/2016/08/201608291034_607214_2984502_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/08/201608291034_607215_2984502_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/08/201608291034_607216_2984502_3.png

抗菌性PSf-PDA超滤膜的制备及表征1引言 PSf超滤膜在使用中不能杀死滤前水中的细菌,导致细菌在PSf膜表面粘附,从而造成微生物聚集,会在PSf膜的表面造成生物污染,引起膜通量下降,阻碍进一步工业应用。制备的亲水性PSf-PDA超滤膜,本章利用PDA的粘性,对PSf膜进行二次修饰,在膜表面继续负载聚六亚甲基胍(PHMG)和纳米银(nAg)两种抗菌剂。2实验部分2.1 实验药品和仪器 实验药品如表1所示:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091714580562_01_2648817_3.jpg实验仪器如表2所示:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091714585086_01_2648817_3.jpg2.2抗菌性PSf-PDA超滤膜的制备http://ng1.17img.cn/bbsfiles/images/2017/10/2015091714594835_01_2648817_3.jpg 取PSf超滤膜,剪成小片,放在多巴胺(DA,2 g·L-1)Tris溶液(0.01 mol·L-1, pH=8.5)中浸泡3 h,然后在4 g·L-1的mPEG-NH2溶液浸泡1h,然后将膜用超纯水洗净,然后分别在2%的PHMG水溶液或者AgNO3溶液浸泡,1 h后取出来用超纯水洗净保存,进行结构和性能的表征。涂覆过程如图1所示。2.3 抗菌性实验及评价2.3.1生物试剂的配制 PBS缓冲液的配制:磷酸二氢钾(KH2PO4)0.24g,磷酸氢二钠(Na2HPO4)1.44g,氯化钠(NaCl)8.0g,氯化钾(KCl)0.2g,加水至1000mL,调节pH 为 7.4。 LB液体培养基:将25g干粉型LB培养基(主要成分为蛋白胨10 g、酵母5 g和氯化钠10 g)溶解到100mL超纯水中,转移到1L的容量瓶中,定容到1L,并保存。 ECC显色培养基:取3.28g瓶内干粉,用100mL超纯水溶解,加热至100℃,并不断搅拌,2 min后大量气泡产生,等到琼脂完全溶解后,将培养基倾入无菌培养皿中,冷却至室温,变成固体后冷藏备用。 琼脂培养基:将3.3 g营养琼脂溶解到100 mL超纯水中,加热煮沸,等待琼脂完全溶解,将培养基倾入无菌培养皿中,凝固后冷温保存。2.3.2大肠杆菌的培养 将在4℃冰箱中保存下的大肠杆菌菌液取出,放在37℃恒温培养箱里放置活化0.5 h,然后在无菌室里接种到50mL的 LB液体培养基里,放在在 37℃恒温培养箱里培养基中剩余的大分子等生长介质,最后将大肠杆菌溶液稀释106倍待用。2.3.3 抑菌实验 抑菌环法:本实验利用抑菌环来判定膜表面的nAg颗粒和膜表面的PHMG的抑菌效果。将上面的大肠杆菌接种到上述的LB培养基中,37℃下培养24 h,将用AgNO3溶液和PHMG溶液浸泡过的PSf-PDA超滤膜剪成小圆片,用UV灯照射30min灭菌。然后将他们放在已涂布的琼脂培养基中,观察抑菌环的情况。 平板菌落计数法:将大肠杆菌溶液加到超滤杯中,将用AgNO3溶液和PHMG溶液浸泡过的PSf-PDA超滤膜过滤一段时间,然后将过滤后的膜放到ECC显色培养基表面铺平,然后放到37℃培养箱中培养24 h,观察膜表面菌落生长的情况。3. 结果与讨论3.1 抗菌性PSf-PDA复合超滤膜制备的优化3.1.1 AgNO3涂覆浓度对膜性能的影响 选用不同浓度(50ppm、500ppm、5000ppm)的硝酸银溶液对PSf-PDA复合膜进行涂覆,时间为1h,并测定接触角和膜阻力的结果。膜阻力结果如图2所示,结果表明随着涂覆AgNO3溶液浓度的升高,膜阻力逐渐增大,从50 ppm时的2.22×109m-1升高到5000ppm时的2.39×109 m-1,是因为随着nAg颗粒在膜表面的形成,导致膜孔在一定程度上发生堵塞,造成膜阻力升高。接触角结果如图3所示。结果表明,随着涂覆AgNO3溶液浓度的升高,膜表面的接触角逐渐变大,说明亲水性逐渐降低,这跟膜表面形成的nAg颗粒的疏水性有关,涂覆的nAg颗粒越多,膜表面的亲水性越低。http://ng1.17img.cn/bbsfiles/images/2017/10/2015091715034293_01_2648817_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509171503_566443_2984502_3.jpg3.1.2 AgNO3涂覆时间对膜性能的影响 选用不同时间(1 h、2 h、3 h)的AgNO3溶液对PSf-PDA复合膜进行涂覆,浓度为500ppm,并测定接触角和膜阻力的结果。膜阻力结果如图4所示,结果表明随着涂覆AgNO3溶液时间的增长,膜阻力逐渐增大,从涂覆1 h时的2.26×109m-1到涂覆3h的2.66×109 m-1,是因为随着nAg颗粒在膜表面的形成,导致膜孔在一定程度上发生堵塞,造成膜阻力升高。接触角结果如图5所示。结果表明,随着涂覆AgNO3溶液时间的增长,膜表面的接触角逐渐变大,说明亲水性逐渐降低,这跟膜表面形成的纳米银涂覆层的疏水性有关,涂覆的纳米银越多,膜表面的亲水性越小。http://ng1.17img.cn/bbsfiles/images/2015/09/201509171506_566445_2984502_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509171506_566446_2984502_3.jpg 随着AgNO3浓度的增加,膜表面的nAg颗粒会使膜阻力逐渐增大,膜表面的接触角增大,亲水性降低,最佳的涂.覆浓.度为500 mg·L-1。 随着AgNO3涂覆时间的增加,膜表面的nAg颗粒会使膜阻力逐渐增大,膜通量逐渐减小,膜表面的接触角增大,亲水性降低,最佳的涂覆时间为1 h。
维生素D为何要用正相半制备色谱净化再用反相色谱定量?可操作性强吗?如果改用硅胶柱净化而不用半制备色谱行不行?食品中的维生素不太好做,有哪位高手经验丰富,望赐教。另外,一般食品中添加的维生素A是什么形态的,我们买的标样是乙酸酯,跟样品的保留时间不同。而我从另一个实验室找的维生素A标样就和样品保留时间一致,但是有两个峰,真搞不懂了。
青龙衣green walnut husks来源于胡桃科胡桃属植物胡桃和胡桃楸的未成熟果实的干燥外果皮。胡桃醌是青龙衣中的萘醌类化合物,也是主要活性成分[1]。现代药理研究表明,胡桃醌具有抗炎、抗菌、抗肿瘤等作用[2-5],对多种肿瘤细胞增殖均有抑制作用。目前,已证实胡桃醌能抑制宫颈癌细胞生长,并诱导其凋亡、抑制细胞迁移、侵袭[6-7]。其对肝癌HepG2细胞的体内外抑制活性显著,能够上调死亡受体5(death receptor 5,DR5)表达,通过ROS介导的p53信号通路激活,促进自噬体形成,诱导细胞的凋亡与自噬[8]。胡桃醌对人乳腺癌MCF-7细胞抑制生长效果明显,与时间和浓度呈正相关,同时使Bcl-2相关X蛋白/B淋巴细胞瘤-2(Bcl-2 associated X protein/B-cell lymphoma-2,Bax/Bcl-2)比值升高,半胱氨酸天冬氨酸蛋白酶-3(cystein- asparate protease-3,Caspase-3)、Caspase-9被激活,诱导细胞凋亡[9]。但胡桃醌水溶性差,易升华,能随水蒸汽挥发,长期存放易发生氧化分解,限制了其在新药开发和在临床上的应用[10],因此,针对其药理活性及潜在应用,设计一种可有效提高胡桃醌稳定性的递药体系具有重要意义。 两亲性嵌段共聚物是在自组装过程中将疏水性药物包覆或键合在聚合物中形成的载药纳米胶束,其能够弥补传统药物水溶性差、吸收率低等不足,可提高药物生物利用度,实现靶向控制释放,在抗癌药物递送中被广泛应用[11]。白及多糖(Bletilla striata polysaccharide,BSP)是从兰科白及属植物白及Bletilla striata (Thunb.) Reichb. f.的干燥块茎中提取得到的一类水溶性多糖,作为天然高分子材料,具有结构稳定、生物可降解、生物安全性高、易于修饰改造等特点,逐渐成为一种纳米药物递送系统的新型优良载体材料[12]。维生素E琥珀酸酯(vitamin E succinate,VES)是维生素E的类似物,因具有较长的脂肪链而疏水性较强,将其和白及多糖连接可提高包载药物的稳定性。VES还能够抑制肿瘤细胞生长和诱导肿瘤细胞凋亡,且只对肿瘤细胞有抑制作用,对正常的组织细胞无任何不良反应,因此VES具有药物和载体的双重作用[13-14],在递送药物的同时达到辅助治疗的效果。 本实验以白及多糖为亲水端,VES为疏水端,合成两亲性嵌段共聚物BSP-VES,将其作为载体制备胡桃醌载药胶束(Jug/BSP-VES),同时考察制备过程中各因素对包封率和载药量的影响,采用星点设计-效应面法(central composite design-response surface methodology,CCD-RSM)优化Jug/BSP-VES胶束的处方和工艺,并进行质量评价,为传统中药青龙衣及其活性成分胡桃醌的开发及临床应用提供参考。 1 仪器与材料 1.1 仪器 Agilent 1260 Series型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],美国安捷伦有限公司;DF-101S型集热式恒温加热磁力搅拌器,上海秋佐科学仪器有限公司;KQ-200KDB型超声波清洗器,昆山市超声仪器有限公司;UV-765型紫外-可见分光光度计,上海精密科学仪器有限公司;Advantage型台式托盘冻干机,美国VirTis公司;80-2型电动离心机,上海浦东物理光学仪器厂;Zetasizer Nano ZSE型纳米粒度电位仪,英国马尔文公司;FTIR-650型傅里叶变换红外光谱仪,天津港东科技股份有限公司;970CRT型荧光分光光度计,北京恒奥德仪器有限公司;Hula Dancer Digital型涡旋混合器,德国IKA公司;Talos F200S G2型透射电子显微镜(TEM),赛默飞仪器公司。 1.2 试药 胡桃醌原料药(批号A2007171,质量分数≥97%)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC)、4-二甲氨基吡啶(DMAP),上海阿拉丁试剂有限公司;白及多糖,批号GH210721,西安国豪生物科技有限公司;胡桃醌对照品,批号RFS-H07511804026,质量分数>98%,成都瑞芬思生物科技有限公司;VES,批号VS1210200734,西安海斯夫生物科技有限公司;芘,分析纯,上海九鼎化学有限公司。 2 方法与结果 2.1 BSP-VES聚合物的合成 称取3.2 g BSP超声溶解于30 mL DMSO中。另称取适量VES、DMAP和EDC(nVES∶nDMAP∶nEDC=1∶1∶1.2)溶于DMSO后磁力搅拌活化1 h,BSP溶液缓慢滴入,密封圆底烧瓶,38 ℃水浴搅拌下反应48 h,室温冷却后移至透析袋(截留相对分子质量3 500)中用纯化水透析2 d以除去未反应试剂。将溶液3 500 r/min离心(离心半径10 cm)15 min取上清液,?20 ℃冰箱中预冻,随后进行冷冻干燥,得到棕色絮状疏松固体,置于4 ℃冰箱中冷藏备用,反应式见图1。 图片 2.2 BSP-VES的表征及结果 2.2.1 核磁共振氢谱(1H-NMR)检测 以D2O为溶剂,对BSP-VES合成产物进行1H-NMR分析。结果如图2所示,δ 3.0~4.0处宽峰为白及多糖上甘露糖和葡萄糖单元中的亚甲基和次甲基(CH2-O和CH-O)的质子峰,δ 0.8~1.0附近为VES中甲基(e)、亚甲基信号峰,δ 5.31处为白及多糖(1,6)糖苷键(a)的质子化学位移。以上结果表明合成产物为BSP-VES[15]。 图片 2.2.2 红外光谱(IR)检测 采用IR法分别对BSP、VES、BSP-VES进行表征,红外扫描范围为4 000~500 cm?1,结果如图3所示。BSP的结果图(图3-a)中,3 384.56、2 921.63 cm?1为O-H和C-H的伸缩振动峰,1 149.37、1 076.08、1 025.94 cm?1为吡喃糖苷构型的特征峰。VES的结果图(图3-b)中,2 923.56 cm?1为-CH2、-CH的伸缩振动峰,1 749.12、1 710.55 cm?1为羧基和酯基中C=O伸缩振动峰,1 373.07、1 157.08 cm?1为-CH3和C-O的伸缩振动峰。BSP-VES的结果图(图3-c),其中2 921.63 cm?1处的C-H伸缩振动峰增强,说明有VES中大量-CH2、-CH3的引入,1 739.48 cm?1为酯基中C=O伸缩振动峰,1 567.84 cm?1为VES中苯环骨架振动峰,揭示了VES的引入[16]。 图片 2.3 Jug/BSP-VES胶束的制备 采用溶剂挥发法制备Jug/BSP-VES胶束[17]。称取20 mg的BSP-VES于15 mL水中,称取2 mg胡桃醌溶于3 mL无水乙醇中,在搅拌下将含药溶液滴加至水相中,在30 ℃下搅拌6 h,有机溶剂挥发完全后即得Jug/BSP-VES胶束溶液。预冻后,置于冻干机中,取出即得冻干粉。 2.4 Jug/BSP-VES中胡桃醌含量测定方法 2.4.1 色谱条件 色谱柱为依利特Kromasil(250 mm×4.6 mm,5 μm);流动相为甲醇-水(70∶30);检测波长248 nm;柱温25 ℃;体积流量1.0 mL/min;进样量10 μL。 2.4.2 溶液的配制 (1)对照品溶液的配制:精密称取胡桃醌对照品5.0 mg,置于25 mL量瓶中,甲醇溶解并定容,得质量浓度为200 μg/mL的对照品储备液。 (2)供试品溶液的配制:精密吸取Jug/BSP-VES胶束溶液0.5 mL至10 mL量瓶中,甲醇破乳并定容至刻度,摇匀,即得Jug/BSP-VES供试品溶液。空白胶束供试品溶液同法操作。 2.4.3 专属性考察 分别取适量空白胶束供试液、适当浓度的胡桃醌对照品溶液及Jug/BSP-VES供试品溶液各10 μL,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],按“2.4.1”项下色谱条件测定,记录色谱图。结果见图4,空白胶束在胡桃醌处无干扰,专属性良好。 图片 2.4.4 线性关系考察 取“2.4.2”项下对照品溶液适量,加甲醇稀释,得到系列质量浓度为1、5、10、30、50、70、100 μg/mL的对照品溶液,按“2.4.1”项下色谱条件进样测定,记录峰面积,以峰面积(Y)对质量浓度(X)进行线性方程拟合,得回归方程为Y=44.786 X-38.423,r=0.999 8,结果表明胡桃醌在1~100 μg/mL线性关系良好。 2.4.5 精密度试验 取“2.4.4”项下低、中、高3个质量浓度(分别为5、30、70 μg/mL)胡桃醌对照品溶液,同1 d内各质量浓度分别进样5次,计算日内精密度;各质量浓度连续进样5 d,计算日间精密度。日内与日间精密度RSD均小于2.0%,表明仪器的精密度良好。 2.4.6 稳定性试验 精密吸取同一供试品溶液在0、2、4、8、12、24 h下,按照“2.4.1”项下色谱条件进行测定,结果峰面积的RSD值为0.596%,表明供试品溶液在24 h内稳定性良好。 2.4.7 重复性试验 取同一批Jug/BSP-VES 6份,按“2.4.2”项方法制备供试品溶液,按照“2.4.1”项下色谱条件进行测定,计算胡桃醌质量浓度的RSD值为1.03%,表明测定方法的重复性良好。 2.4.8 加样回收率试验 精密量取200 μg/mL胡桃醌对照品溶液0.25、1.50、3.50 mL各3份于10 mL量瓶中,加入BSP-VES聚合物,用甲醇定容,分别得到胡桃醌质量浓度为5、30、70 μg/mL的溶液,按“2.4.1”项下色谱条件测定胡桃醌的含量,测得加样回收率均在99%~102%,RSD均小于2.0%,表明检测结果准确可靠。 2.5 胡桃醌包封率、载药量的测定 采用离心法进行聚合物胶束药物包封率和载药量的测定[18]。精密吸取Jug/BSP-VES胶束溶液1 mL至1.5 mL离心管中,3 000 r/min离心(离心半径8 cm)10 min,除去游离药物,吸取0.5 mL上清液,甲醇破乳并定容至刻度,摇匀,按“2.4.1”项下色谱条件进样分析。另取Jug/BSP-VES胶束溶液0.5 mL至10 mL量瓶中,甲醇破乳并定容至刻度,摇匀,按“2.4.1”项下色谱条件进样分析。将所得峰面积带入线性方程计算胡桃醌的包封率和载药量。 包封率=W胶束中药物量/W总药量 载药量=W胶束中药物量/W胶束质量 2.6 单因素考察 2.6.1 有机溶剂种类考察 固定其他条件不变,即有机溶剂用量为3 mL,挥发时间为6 h,制备温度为30 ℃,载药比为10∶1,水相用量为15 mL,分别加入有机溶剂氯仿、丙酮、甲醇、无水乙醇,考察不同有机溶剂种类对载药量和包封率的影响。结果(表1)显示,以无水乙醇为溶剂时,制备的胶束溶液包封率和载药量最高,因此,选择无水乙醇作为溶剂来制备Jug/BSP-VES胶束。 图片 2.6.2 有机溶剂用量考察 固定其他条件不变,即有机溶剂为无水乙醇,挥发时间为6 h,制备温度为30 ℃,载药比为10,水相用量为15 mL,加入一定量的BSP-VES和胡桃醌分别溶解于1、2、3、4、5 mL无水乙醇中,考察不同有机溶剂用量对载药量和包封率的影响。结果(表2)显示,当有机溶剂用量为3 mL时胡桃醌的载药量和包封率最高,因此,选择3 mL作为有机溶剂用量。 图片 2.6.3 挥发时间考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,制备温度为30 ℃,载药比为10,水相用量为15 mL,考察挥发时间在4、5、6、7、8 h时,不同挥发时间对载药量和包封率的影响。结果(表3)显示,当挥发时间为6 h时胡桃醌的载药量和包封率最高,因此,选择6 h作为挥发时间来制备Jug/BSP-VES胶束。 图片 2.6.4 制备温度考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,载药比为10,水相用量为15 mL,考察制备温度在25、30、35、40、45 ℃时,不同制备温度对载药量和包封率的影响。结果(表4)显示,随着制备温度的增加,胡桃醌的载药量与包封率先升高后降低,因此将25~35 ℃的制备温度作为待优化项进行CCD-RSM实验。 图片 2.6.5 载药比考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,制备温度为30 ℃,水相用量为15 mL,精密称取药物2 mg,加入不同质量的载体,即载药比分别为6、8、10、12、14时,考察不同载药比对载药量和包封率的影响。结果(表5)显示,随着载体量的增加,胡桃醌的包封率先升高后降低,因此将8、10、12的载药比作为待优化项进行CCD-RSM实验。 图片 2.6.6 水相用量考察 固定其他条件不变,即有机溶剂为无水乙醇,用量为3 mL,挥发时间为6 h,制备温度为30 ℃,载药比为10,考察水相用量在5、10、15、20、25 mL时,不同水相用量对载药量和包封率的影响。结果(表6)显示,随着水相用量的增加,胡桃醌的载药量与包封率先升高后降低,因此将10~20 mL的水相用量作为待优化项进行CCD-RSM实验。 图片 2.7 CCD-RSM优化处方 在单因素考察实验基础上,进一步采用CCD- RSM优化制剂工艺。选取载药比(X1)、水相体积(X2)、制备温度(X3)3个因素,每个因素设定5个水平(?1.682、?1、0、+1、+1.682)。以胡桃醌包封率(Y1)和胡桃醌载药量(Y2)为考察指标进行3因素,5水平的CCD-RSM实验,结果见表7。采用Design-Expert统计软件对表7数据进行统计处理,并获得Y1、Y2值对自变量X1、X2、X3的多元线性回归方程,各考察指标的2项式拟合方程如下Y1=90.010+0.165 9 X1+0.700 4 X2-0.1071 X3-0.656 4 X1X2-1.020 X1X3-0.516 1 X2X3-4.430 X12-3.520 X22-4.100 X32;Y2=6.430-0.408 3 X1+0.288 5 X2-0.210 6 X3+0.251 8 X1X2-0.380 1 X1X3-0.374 7 X2X3-0.004 7 X12-0.057 7 X22-0.111 9 X32。各方程的方差分析结果见表8,结果表明该模型与实际试验拟合程度良好,且各因素影响显著用该模型分析和预测胶束的制备工艺是合适的。 图片 图片 利用Design-Expert统计软件绘制自变量对因变量的效应面和等高线图,结果见图5。最终确定最佳条件范围得到的最优处方:BSP-VES与胡桃醌的投药量分别为20 mg和2 mg,水相用量15 mL,制备温度30 ℃。预测在此条件下制备Jug/BSP-VES的包封率和载药量分别为90.047%、6.559%。 图片 2.8 最优处方的验证试验 按最优处方平行制备3批Jug/BSP-VES胶束溶液,测定其中胡桃醌的包封率、载药量。胡桃醌的平均包封率为(88.44±1.24)%、RSD值为1.79%,胡桃醌平均载药量为(6.54±0.02)%、RSD值为1.90%,RSD值均<3%,表明模型预测可靠,工艺重现性较好。 2.9 Jug/BSP-VES胶束的表征 2.9.1 Jug/BSP-VES胶束溶液外观及形态观察 取制备好的Jug/BSP-VES溶液,观察外观及丁达尔现象;取适量Jug/BSP-VES溶液纯水稀释,滴加至专用铜网上,待风干后,通过透射电子显微镜(TEM)观察形态并拍照。结果如图6所示,Jug/BSP-VES胶束溶液为黄色澄清溶液,丁达尔效应明显;在TEM下观察到Jug/BSP-VES胶束呈类球形,分散均匀。 图片 2.9.2 BSP-VES临界聚集浓度(critical aggregation concentration,CAC)的测定 采用芘荧光探针法检测聚合物的CAC。配制质量浓度为1 mg/mL的芘溶液和1 mg/mL的BSP-VES母液。取9个西林瓶,各加入0.25 mL芘溶液,氮气吹干后各加入不同质量浓度的1 mL BSP-VES溶液。稀释后BSP-VES溶液的质量浓度分别为100.00、50.00、10.00、5.00、1.00、0.50、0.10、0.05、0.01 μg/mL。涡旋5 min后超声30 min,室温避光静置24 h。荧光分光光度计的激发波长为330 nm,测定各溶液中芘的荧光吸收,以373、384 nm处样品的荧光光度值之比(I373/I384)对质量浓度的对数作图,两条切线的交点为CAC值。结果如图7所示,当BSP-VES质量浓度较低时,I373/I384值较小,当BSP-VES质量浓度增大时,I373/I384值增大,取图中两直线相交处为BSP-VES的CAC值,经计算,CAC值为5.95 μg/mL。 图片 2.9.3 包封率和载药量的测定 按最优处方制备Jug/BSP-VES胶束溶液,测定其包封率和载药量,方法同“2.5”项。结果发现Jug/BSP-VES胶束溶液的包封率为(89.140±1.163)%(n=3),载药量为(6.493±0.087)%(n=3)。 2.9.4 粒径及ζ电位测定 按最优处方制备Jug/ BSP-VES胶束溶液,Zetasizer Nano ZSE纳米粒度电位仪测定其粒径、粒度分布及ζ电位。结果如图8所示,测得Jug/BSP-VES胶束溶液的平均粒径为(120.30±2.80)nm,PDI为0.169±0.014,ζ电位为(?27.00±1.25)mV。 图片 2.9.5 差示扫描量热法(differential scanning calorimetry,DSC) 分别称取适量胡桃醌、BSP- VES、胡桃醌原料药物理混合物和Jug/BSP-VES胶束样品置于铝制样品盘中压制,氮气为保护气,扫描范围25~350 ℃,加热速率10 ℃/min。结果如图9所示。胡桃醌的特征吸收峰在156 ℃,BSP-VES的特征吸收峰为184 ℃,与胡桃醌的特征峰不重叠;物理混合物中,二者特征峰均出现,而Jug/ BSP-VES胶束的热量曲线上无胡桃醌的特征峰,说明胡桃醌已被成功包载进载体,特征吸收峰消失。 图片 2.9.6 储存稳定性考察 按最优处方制备Jug/ BSP-VES胶束溶液,在pH 4.5,4 ℃和25 ℃条件下测定其在第1、3、7、15 d的粒径和包封率。结果如表9所示,在4 ℃下,Jug/BSP-VES的粒径和包封率无较大变化,说明储存稳定性较好;在25 ℃下储存效果相对较差,随时间增加,胶束溶液粒径变大,包封率降低,因此4 ℃为Jug/BSP-VES胶束溶液的最优储存条件。 图片 2.9.7 体外释放考察 采用透析法考察胡桃醌和Jug/BSP-VES胶束溶液的体外释药情况。分别将胡桃醌、Jug/BSP-VES胶束溶液置于透析袋(截留相对分子质量3 500)中,透析袋两端夹紧,分别浸没在含有0.5%聚山梨酯-80的醋酸-醋酸钠缓冲液(Ph 4.5)中;恒温水浴(37.0±0.5)℃,转速100 r/min,每组平行进行3组试验,分别于选定的时间点收集5 mL样品,收集后补加等量同温的释放介质,所得到的样品经微孔滤膜滤过后进行HPLC分析,体外释药曲线如图10所示。胡桃醌溶液在6 h时释放到80%左右,Jug/BSP-VES胶束在48 h时的释放率为(82.13±2.51)%,达到了明显的缓释作用,表明将原料药制备成胶束可减缓药物的释放速度。 图片 3 讨论 胡桃醌作为抗肿瘤活性成分具有一定的毒性,对金鱼的半数致死量(median lethal dose,LD50)为1.3 mg/L,对小鼠ig给药、ip的LD50值分别为2.5、25.0 mg/kg[19-20]。此外,胡桃醌及其代谢产物能与肾脏细胞溶质蛋白共价结合,造成肾脏毒性[21]。研究表明,酒精能使胡桃醌中的毒性成分转变为其他物质[22],以酒精作为溶剂的胡桃醌制剂通常不显毒性。本研究通过BSP与VES发生酯化反应成功制备了BSP-VES胶束,以胡桃醌为模型药物,通过溶剂挥发法制备了Jug/BSP-VES载药胶束。Jug/ BSP-VES载药胶束外观呈类球型,粒度测定结果显示,Jug/BSP-VES胶束溶液的粒径图显示峰形呈单峰,分布范围较窄,说明胶束溶液粒径均一。TEM下观察到的Jug/BSP-VES胶束,其粒径比粒度仪测定结果较小,可能是由于在制样过程中胶束水分的挥干导致粒子发生皱缩所致。BSP-VES作为两亲性高分子材料,在水相中的浓度超过临界胶束浓度后可形成胶束,制备方法简便。 本实验设计了一种可提高胡桃醌稳定性的载药胶束,拟制成温敏凝胶剂、采用阴道给药的方式,用于治疗阴道炎症、宫颈癌术后等。正常人体阴道pH值范围在3.5~4.8[23],因此,体外释放实验采用的是pH 4.5并含有0.5%聚山梨酯-80的醋酸-醋酸盐缓冲液[24],来模拟阴道中的酸性环境。在稳定性研究中,也重点考察了上述条件下载药胶束的储存稳定性,而并未采用通常的PBS(0.01 mol/L,pH 7.4)缓冲体系和含10% FBS的PBS(0.01 mol/L,pH 7.4)缓冲体系。另外,本实验所制备的Jug/BSP-VES载药胶束处方中尽可能减少了辅料种类,以避免腔道用药过程中的副作用及不良反应。 在单因素实验中,本实验考察各因素对处方工艺的影响。制备温度的高低主要影响有机溶剂除去的速度,温度过高或过低,引起有机溶剂挥发速度过快或过慢,均不利于胶束对药物的包载[25]。因考虑到温度对制备的影响较大,在25~35 ℃时Jug/ BSP-VES胶束中的胡桃醌含量不稳定,因此,对制备温度作进一步实验。 对有机溶剂用量的考察中,有机溶剂用量过少时,容易造成药物不能完全溶解,随着有机溶剂用量的增加,药物在溶剂中均匀分散,能与胶束较好地结合,当有机溶剂用量过多时,在有限的时间内,容易造成挥发不完全导致包封率降低[25],因此选择3 mL作为有机溶剂用量。 综上所述,本研究制备的Jug/BSP-VES胶束,通过单因素实验与CCD-RSM优化后,包封率好,粒径均一,稳定性良好,为胡桃醌制剂的应用开发奠定了基础。
【序号】:1【作者】: 王先津代元坤苑志磊贺继东【题名】:丁二酸酐酰化壳聚糖-g-PHB纳米微球的制备与表征【期刊】:化学与生物工程. 【年、卷、期、起止页码】:2018,35(07)【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C44YLTlOAiTRKibYlV5Vjs7i0-kJR0HYBJ80QN9L51zrPwrSRYDLTFoseLVM05aDgmt1NJw4N2j08qxaqTe_5CqI&uniplatform=NZKPT
【序号】:1【作者】: 王丰艳【题名】:载银聚乙烯醇/羧甲基壳聚糖/海藻酸钠水凝胶伤口敷料的制备及性能表征【期刊】:中国矿业大学【年、卷、期、起止页码】:2020【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD202101&filename=1020657862.nh&uniplatform=NZKPT&v=ZF5BfbXAmb_dlc4cwqxnD_msfmT0mginc4LZIpzNs5PPk1YtgbBxuWpu40koFpPG
![SiO2-[P888Allyl]Br色谱固定相的制备及表征](https://ng1.17img.cn/bbsfiles/images/2022/11/202211162155248793_4119_5389809_3.png)
[align=left][font='times new roman'][size=16px] SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-[P[/size][/font][font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font][font='times new roman'][size=16px]]Br[/size][/font][font='times new roman'][size=16px]色谱固定相的制备[/size][/font][font='times new roman'][size=16px]及[/size][/font][font='times new roman'][size=16px]表征[/size][/font][/align][align=left]SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相的制备分三步进行。[/align][align=left]球形多孔硅胶在100℃下干燥24 h,备用。[/align]第一步为离子液体的制备:通过亲核取代反应制备季膦离子液体[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br。首先,在0℃条件下,将6.8 mL二氯甲烷置于25 mL圆底烧瓶中,在冰浴条件下迅速加入0.3 mL烯丙基溴和1.0 mL三辛基膦,将烧瓶置于室温,N[font='times new roman'][sub][size=16px]2[/size][/sub][/font]氛围下搅拌50 min,时刻注意反应情况,避免胶塞漏气。27℃下蒸发二氯甲烷,得到透明浅黄色粘稠状液体,正己烷洗涤产物5次,60℃下真空干燥12 h,得到季膦离子液体[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br(1.1 g,87.7%)。第二步为巯丙基化硅球(SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS)的制备:先要称取2.5 g干燥的球形多孔硅胶置于250 mL圆底烧瓶底部,加35 mL无水甲苯使其重悬,之后向反应体系中加入7.0 mL MPS。混合均匀后充入N[font='times new roman'][sub][size=16px]2[/size][/sub][/font],110℃下加热回流24 h。反应结束后,用甲苯和大量的甲醇洗涤产物以充分洗去反应体系中的杂质,抽提。90℃下干燥过夜,得到SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS。第三步为SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br固定相的制备:通过“巯基-烯”点击化学反应制备SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br固定相。称取2.3 g SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS置于250 mL圆底烧瓶中;称取5.6 g [P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br溶解于40 mL甲醇中,摇匀后加入250 mL圆底烧瓶中;继续加入0.06 g AIBN,三者均匀混合后,N[font='times new roman'][sub][size=16px]2[/size][/sub][/font]氛围下65℃下加热回流反应24 h。反应结束后用大量甲醇洗涤产物4次,以充分洗去反应体系中的杂质,抽提。90℃下干燥24 h,得到SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相。[align=center][/align][align=left][font='times new roman'][size=16px]SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-[P[/size][/font][font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font][font='times new roman'][size=16px]]Br[/size][/font][font='times new roman'][size=16px]色谱柱的填充[/size][/font][/align]采用高压匀浆法装SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱柱。称取2.0 g SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br固定相,将SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱填料与匀浆液混合,超声处理两秒使其均匀分散,匀浆液按照氯仿-环己醇(2:3,v/v)比例进行配置。将混合完全的溶液倒入至已经连接好空柱管(150 mm × 4.6 mm)的匀浆罐中,连接好装置避免漏液。调节压力上升至35 MPa,加压液甲醇-异丙醇(1:1,v/v)会随着管路流过匀浆罐下方的色谱柱,在此压力下维持30 min,降低压力为0 MPa,静置30 min。将柱管卸下后装入筛板和柱头后,用乙腈冲洗色谱柱,备用。[align=left][font='times new roman'][size=16px]SiO[/size][/font][font='times new roman'][sub][size=16px]2[/size][/sub][/font][font='times new roman'][size=16px]-[P[/size][/font][font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font][font='times new roman'][size=16px]]Br[/size][/font][font='times new roman'][size=16px]色谱固定相表征[/size][/font][/align]通过核磁共振波谱仪和质谱仪对合成的离子液体[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br进行了表征,如图所示,可验证离子液体[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br已成功合成。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211162155248793_4119_5389809_3.png[/img][/align][align=center][size=13px]图[/size][size=13px] [P[/size][font='times new roman'][sub][size=13px]888Allyl[/size][/sub][/font][size=13px]]Br[/size][size=13px]的核磁共振谱图和质谱图[/size][/align]硅球和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相中的C和H元素通过元素分析仪进行测试分析。如图2-3a所示,空硅球中C和H元素的含量分别为0.46%和0.72%,而SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相中C和H元素含量分别增加到4.3%和1.4%。采用扫描电子显微镜对硅球和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br材料进行表面形态表征。如图2-3b所示,与SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]相比,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相表面更加粗糙,可以观察到明显突起,可初步证明SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相已经成功制备。硅球、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相采用傅里叶变换红外光谱仪对其进行表征。图2-3c中可以明显看到在硅球、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br的红外谱图中1100 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]处的强吸收峰,SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br是以二氧化硅为基底,所以此处峰可以认定为Si-O的伸缩振动所引起的。硅球中存在硅醇基团,其伸缩振动特征峰可以归于SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]谱图中980 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]处。在SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS谱图中,由于980 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]处峰的消失,可以认定为硅球中的硅醇基团都被MPS掩盖,证明MPS已经成功键合到SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]上。相比于SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS,可以在SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br固定相中观察到2930 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]和2850 cm[font='times new roman'][sup][size=16px]-1[/size][/sup][/font]两处吸收峰,这属于[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br结构中典型的甲基和亚甲基的特征峰。最后,通过热重分析仪对硅球、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相的稳定性进行表征。如图2-3d所示,在100℃以内硅球、SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br仍然出现了极少量质量损失,这可能是材料内部存在少量的结合水或表面吸附了空气中的水分子造成的。当温度超过100℃,水分子到达沸点后挥发。SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-MPS和SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br分别在400℃和300℃质量损失迅速下降,这是由于材料表面键合的有机功能团脱落导致的。综合上述分析,可以证明SiO[font='times new roman'][sub][size=16px]2[/size][/sub][/font]-[P[font='times new roman'][sub][size=16px]888Allyl[/size][/sub][/font]]Br色谱固定相的成功制备。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211162155265798_7481_5389809_3.png[/img][/align][align=center][size=13px]图[/size][size=13px] [/size][size=13px]硅球[/size][font='宋体'][size=13px]、[/size][/font][size=13px]SiO[/size][font='times new roman'][sub][size=13px]2[/size][/sub][/font][size=13px]-MPS[/size][size=13px]和[/size][size=13px]SiO[/size][font='times new roman'][sub][size=13px]2[/size][/sub][/font][size=13px]-[P[/size][font='times new roman'][sub][size=13px]888Allyl[/size][/sub][/font][size=13px]]Br[/size][size=13px]的元素分析图([/size][size=13px]a[/size][size=13px])[/size][font='宋体'][size=13px]、[/size][/font][size=13px]扫描电镜图([/size][size=13px]b[/size][size=13px])[/size][font='宋体'][size=13px]、[/size][/font][size=13px]红外光谱图([/size][size=13px]c[/size][size=13px])和热重分析图([/size][size=13px]d[/size][size=13px])[/size][/align]
有人用过flash反相制备过柱机吗?具体操作方法是什么呀?







 我要推广仪器
我要推广仪器
 下载APP
下载APP