测定氨基酸用氨基酸分析仪好还是用HPLC好,二者有何异同? 请用过二者的专家释疑?多谢!
液质分析有机相:0.1%甲酸的乙腈 或 5mM甲酸铵的甲醇 二者区别?文献中多用0.1%甲酸的乙腈,为什么?
液相分析中化合物具有紫外吸收,用DAD和ELSD区别在哪里?二者相关性如何?

[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析乙二醇,尝试多次后二者峰依然分不开[img=,690,241]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113270516_2976_1782490_3.jpg!w690x241.jpg[/img][img=,690,341]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113300272_8258_1782490_3.jpg!w690x341.jpg[/img][img=,690,326]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113340193_5542_1782490_3.jpg!w690x326.jpg[/img][img=,690,331]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113378262_41_1782490_3.jpg!w690x331.jpg[/img][img=,690,374]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113419511_5133_1782490_3.jpg!w690x374.jpg[/img][img=,690,351]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051113446332_7232_1782490_3.jpg!w690x351.jpg[/img]
[size=5][b]今天做实验遇到一个问题,就是下面的含量测定,结果计算要求“[/b]根据二者吸收度比值计算[/size][b][size=5]”,请教大家怎么计算?麻烦知道的朋友根据我提供的数据列出公式,万分感谢![/size]【含量测定】[/b] [size=3][font=宋体]取本品适量(约相当于乳酸诺氟沙星64mg),精密称定,置200ml量瓶中,加氢氧化钠液(0.1mol/L)振摇使溶解,用氢氧化钠液(0.1mol/L)稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用氢氧化钠液(0.1mol/L)稀释至刻度,摇匀。照《紫外-可见分光光度法标准操作规程》,在273nm波长处测定吸收度;另取诺氟沙星对照品约50mg,精密称定,照上法同样操作,根据二者吸收度比值计算,并将计算结果乘以1.28,即得。诺氟沙星对照品称取量:52.84mg样品称取量:1.31112g(乳酸诺氟沙星的标示量为100g:5g)对照品吸收度:0.650样品吸收度:0.631[/font][/size]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=42516]电镀的结晶过程[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=42517]电镀铜系列添加剂的研究[/url][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=42518]电镀常见故障的分析和纠正-1[/url]原题:治疗共同分享
http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif如题 ,这东西貌似挺火的 请大家注意是要标准品鉴于玛卡酰胺和玛卡烯目前没有标准品出售,本帖更改为征集二者的化学结构图,最好是立体的哦!
再做红外鉴别时,标准上经常要压个标准品的图或跟标准光谱集比较,然后要求“二者图谱应一致”,俺想问的是:两张图到底怎么个一致才能判定合格呢??
如何分别测试高氯酸和草酸铵中二者的含量,一般的方法只能测定总的胺值,有没有能准确测试二者混合物中各自的含量?
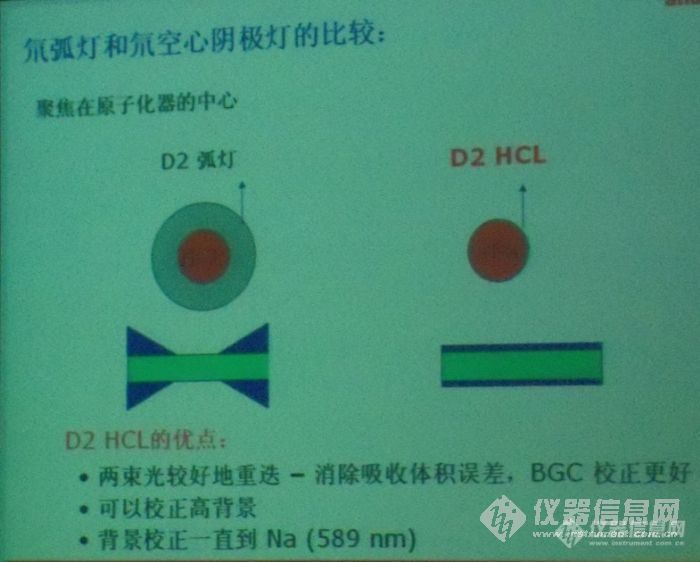
氘弧灯和氘空心阴极灯二者的区别是什么?请老师们做个详细的介绍,谢谢!http://ng1.17img.cn/bbsfiles/images/2012/08/201208032129_381524_2355529_3.jpg
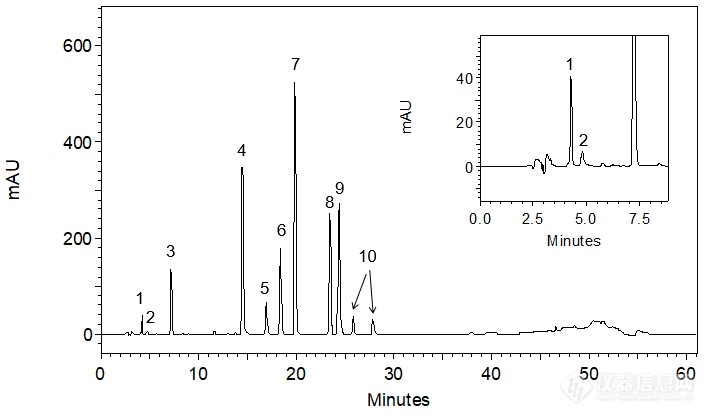
[align=center][b]头孢克洛有关物质——与9种杂质的共同分析[/b][/align]头孢克洛(cefaclor)为白色至微黄色粉末或结晶性粉末的化学品,微臭,本品在水中微溶,在甲醇、乙醇、三氯甲烷或二氯甲烷中几乎不溶,分子式:C15H14ClN3O4S。头孢克洛是β-内酰胺类抗生素,头孢菌素类药,是第二代头孢菌素,主要适用于敏感菌所致的急性咽炎、急性扁桃体炎、中耳炎、支气管炎、肺炎等呼吸道感染、皮肤软组织感染和尿路感染等。[align=center][img=,144,171]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140859582934_5220_2222981_3.gif!w144x171.jpg[/img][/align][align=center]头孢克洛[/align][align=center]M.W.: 367.81[/align]本实验对客户提供的头孢克洛原料药以及9种杂质(杂质A、B、C、D、E,7-ACCA,头孢克洛δ-3异构体,α-苯甘氨酸,苯甘氨酸甲酯盐酸盐)进行分析,希望得到杂质混合对照溶液及供试品溶液中各杂质的良好分离。客户反馈,将流动相磷酸盐体系的pH值由4.0提高到4.5可得到杂质混合对照溶液中7-ACCA和α-苯甘氨酸之间的良好分离,但头孢克洛与其相邻杂质E峰之间分离较难。客户前期使用了CAPCELL PAK C[sub]18 [/sub]MGII S3 4.6 mm i.d. × 250 mm色谱柱进行分析,在此基础上,我们尝试了其他填料的几款色谱柱进行分离尝试,分别为CAPCELL PAK C[sub]18[/sub] AQ(S3& S5)、CAPCELL PAK ADME(金刚烷基)、SUPERIOREX ODS、CAPCELL PAK PFP(五氟苯基)、CAPCELL PAK CN(氰基)。首先,参考客户提供的液相条件,使用高极性色谱柱[b]CAPCELL PAK C[sub]18 [/sub]AQ[/b]对杂质混合对照溶液进行分析尝试;为了得到杂质间的更好分离,粒径选择3 μm,如图1,[color=#2F5496]各杂质间均能得到良好的分离结果,头孢克洛与杂质[/color][color=#2F5496]E[/color][color=#2F5496]的分离度为[/color][color=#2F5496]2.70[/color][color=#2F5496],达到基线分离。[/color][color=#2F5496][/color][align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902184290_9307_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 AQ S3 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 头孢克洛 9. 杂质E [/color]10.杂质D[/align][color=#2F5496][img=,555,311]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902187828_2715_2222981_3.png!w555x311.jpg[/img][/color]进一步分析供试品溶液,如图2,由于样品浓度较高,导致头孢克洛主峰向后展宽,进而将杂质E包于其中。[color=#2F5496][/color][align=center][color=#2F5496][img=,659,441]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140915544228_5404_2222981_3.png!w659x441.jpg[/img][/color][/align][align=center]图2 AQ S3 分析供试品溶液结果[/align][align=center][/align][align=left]为使头孢克洛和杂质E之间得到更好的分离,我们尝试对色谱条件进行调整。[/align][align=left][/align][align=left][b]1.调整柱温[/b][/align][align=left][b][/b]首先对温度进行调整:实验过程中发现柱温对头孢克洛与杂质E的出峰行为有较大影响——当柱温设置为20 ℃时,头孢克洛和杂质E之间能够得到良好分离;将温度提高到30℃时,杂质E向前移动趋势较大。为使杂质E峰出在头孢克洛峰前,避免由于供试品中头孢克洛峰的展宽而使杂质E被包于其内,进一步将柱温提高到40℃,发现头孢克洛与杂质E峰重合;最终,将柱温提高到45℃,此时杂质E峰移至头孢克洛峰前,但未能得到理想的分离结果。[/align][align=left][/align][align=center][img=,659,430]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140916597550_373_2222981_3.png!w659x430.jpg[/img][/align][align=center]图3 不同柱温条件下AQ S3分析杂质混合对照溶液结果[/align][align=center][/align][align=left][b]2.调整流动相[/b][/align][align=left][b][/b][/align][align=left]考虑到提高柱温对色谱柱寿命的影响,仍选择初始使用的20℃,对流动相梯度条件进行调整。在增强整体保留时间的同时,发现[color=#538135]头孢克洛和杂质[/color][color=#538135]E[/color][color=#538135]的出峰顺序发生了颠倒[/color],且[color=#538135]分离良好[/color],进而有效避免了杂质E被包于头孢克洛主峰中的问题;而在主峰后出峰的杂质D与头孢克洛之间分离度亦较高,即使供试品溶液中的头孢克洛峰展宽,也不会出现将杂质D包于其中的问题。[/align][align=left]因此我们在此梯度条件下进一步对供试品溶液进行分析,如图4,头孢克洛与各杂质峰之间均能得到良好的分离结果。[/align][align=left][/align][align=center][img=,679,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140917450308_6331_2222981_3.png!w679x417.jpg[/img][/align][align=center]图4 AQ S3分析杂质混合对照溶液及供试品溶液结果(调整梯度)[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][img=,587,335]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918136074_9375_2222981_3.png!w587x335.jpg[/img][/align][align=left][/align][align=left]为使客户有更多的色谱柱选择,本实验室也尝试使用键合金刚烷基的高极性色谱柱CAPCELL PAK ADME分析杂质混合对照溶液和供试品溶液,如图5,在分析杂质混合对照溶液时,能够得到各组分的良好分离,同时发现杂质E和头孢克洛出峰顺序发生颠倒,但同时也发现头孢克洛峰与其后相邻杂质D峰之间分离度较低(Rs=1.71);因此,如图6,在分析供试品溶液时,由于色谱峰向后展宽,使得杂质D被包于头孢克洛主峰中,未能得到理想分离结果。[/align][align=left][/align][align=center][img=,690,426]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918484278_6616_2222981_3.png!w690x426.jpg[/img][/align][align=center]图5 ADME 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][/align][align=center][img=,689,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918485898_9906_2222981_3.png!w689x417.jpg[/img][/align][align=center]图6 ADME 分析杂质混合对照溶液结果[/align][align=left][img=,585,336]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140919331328_5070_2222981_3.png!w585x336.jpg[/img][/align][align=left][/align][align=left][/align][align=left]之后,我们也尝试使用了CN(氰基柱)和PFP(五氟苯基)以及高碳载量的SUPERIOREX ODS色谱柱,在客户提供的色谱条件下对杂质混合对照溶液进行分析,均未能得到更理想的分离结果。[/align]
拉曼光谱谱图比较各位大侠,小女子这厢有礼了!请问拉曼谱图对物质进行鉴别时,如果样品图谱和对照图谱不尽相同时,怎样比较判断二者是否是同一物质呢?有什么方法或是软件么?谢谢大家!
苄嘧磺隆二氯喹啉酸的图谱,二者出峰顺序我做的和标准怎么相反?请说说你们的意见,谢谢!
希望共同分享-电镀产品的检验验标准[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=64578]检验标准[/url]
如题,您的GC-MS日常分析中是定性多一点还是定量分析多一点呢?主要都做哪些分析工作?在不泄密的前提下欢迎大家畅所欲言!回帖积极,中肯的有积分奖励!
紫外可见分光光度计的检测器,有光电倍增管和硅光二极管两种,这二者有什么优缺点?
[align=center][b]什么是弱电?什么是强电?二者有何区别[/b][/align]水木源华[color=#ff0f1d][b]一、什么是弱电?[/b][/color] 弱电一般是指直流电路或音频、视频线路、网络线路、电话线路,直流电压一般在32V以内。家用电气中的电话、电脑、电视机的信号输入(有线电视线路)、音响设备(输出端线路)等用电器均为弱电电气设备。[b]二、什么是强电?[/b] 强电指电工领域的电力部分。特点是功率大、电流大、频率低,主要考虑损耗小、效率高的问题。和弱电的关系很密切,与“弱电”相对。[b]三、二者有何区别[/b] 强电和弱电主要区别是用途的不同。强电是用作一种动力能源,弱电是用于信息传递。 强电和弱电从概念上讲,一般是容易区别的,主要区别是用途的不同。强电是用作一种动力能源,弱电是用于信息传递。家庭电路分为强电和弱电。在电力系统中,36v以下的电压称为安全电压,1kv以下的电压称为低压,1kv以上的电压称为高压,直接供电给用户的线路称为配电线路,如用户电压为380/220v,则称为低压配电线路,也就是家庭装修中所说的强电。 强电与弱电是相对的概念,从概念上讲,主要区别是用途的不同,而不能单纯的以电压大小来界定两者关系(如果非要指定用电压区分的话,那就把36V(人体安全电压)以上划定为强电, 36V(人体安全电压)以下为划定为弱电。) ,两者既有联系又有区别,一般区分原则是:强电的处理对象是能源(电力),其特点是电压高、电流大、功率大、频率低,主要考虑的问题是减少损耗、提高效率,弱电的处理对象主要是信息,即信息的传送和控制,其特点是电压低、电流小、功率小、频率高,主要考虑的是信息传送的效果问题,如信息传送的保真度、速度、广度、可靠性。[b]它们大致有如下区别:[/b](1)交流频率不同 强电的频率一般是50Hz(赫),称“工频”,意即工业用电的频率:弱电的频率往往是高频或特高频,以KHz(千赫)、MHz(兆赫)计。(2)传输方式不同 强电以输电线路传输,弱电的传输有有线与无线之分。无线电则以电磁波传输。(3)功率、电压及电流大小不同 强电功率以KW(千瓦)、MW(兆瓦)计、电压以V(伏)、KV(千伏)计,电流以A(安)、kA(千安)计;弱电功率以W(瓦)、mW(毫瓦)计,电压以V(伏)、mV(毫伏)计,电流以mA(毫安)、uA(微安)计,因而其电路可以用印刷电路或集成电路构成。[b]四、总结。[/b] 当然,强电中也有高频(数百KHz)与中频设备,但电压较高,电流也较大。又如手电筒与电动剃须刀虽然电压很低,功率及电流很小,仍属强电。由于现代技术的发展,弱电己渗透到强电领域,如电力电子器件、无线遥控等,但这些只能算作强电中的弱电控制部分,它与被控的强电还是不同的。[align=center][color=#888888]来源:电力合伙人[/color][/align]
内标法检测二异丙基萘(7种同分异构体),按方法要求分别精确称取5份不同质量的二异丙基萘(7种同分异构体)配成溶液进行GCMS分析。做标线时遇到了困难,只知道二异丙基萘(7种同分异构体)的质量,不知道各同分异构体的质量,这标线怎么做?工作站能实现这7种同分异构体峰面积加和吗?能的话操作步骤是?请各位牛人赐教!谢谢!
新人报道,各位老师,我是做食品添加剂,果胶的。有机磷农残检测的样品处理有两种,我应该用丙酮那种还是二氯甲烷那种,后者简单,二者有什么区别?请指教
环境保护污染防治及监测与环保指令二者的关系是啥?各自属性有什么不同?包含属性如何?
请教各位大侠,谁知道用于分析甲苯二异氰酸酯同分异构体的方法或色谱柱?请与我联系.多谢!电话:0317-3557630邮箱:hczhangqing@sohu.com
电位滴定法和人工滴定法二者有什么优缺点?那位老师知道?
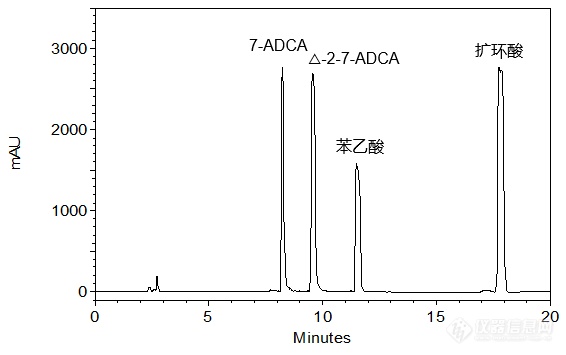
[align=center][b]4种头孢中间体的共同分析[/b][/align][align=right][b]——7-ADCA、△-2-7-ADCA、苯乙酸及扩环酸的分析[/b][/align][align=right][b][/b][/align]客户提供了7-ADCA(7-氨基去乙酰氧基头孢烷酸),△-2-7-ADCA、苯乙酸及扩环酸原料,希望本实验室依据客户所提供的色谱条件筛选合适的C[sub]18[/sub]色谱柱,实现以上4种化合物的稳定良好分析。本实验室参考客户提供的色谱条件,首先尝试使用经过聚合物包被处理的中等极性色谱柱——CAPCELL PAK C[sub]18[/sub] MGII对客户所提供样品进行分析,使用PDA检测器进行检测。如图1,在高浓度大体积进样的情况下,各色谱峰发生严重过载现象,出现平头峰;如图2,降低进样体积至5 μL可得到相对良好峰形,且各组分间能够得到良好分离,分离度均在10.0以上(结果详见表1)。[align=center][img=,566,355]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537444868_8925_2222981_3.png!w566x355.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,575,365]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545063985_5910_2222981_3.png!w575x365.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表1 CAPCELL PAK C[sub]18[/sub] MGII分析结果详表(进样量:5 μL)[/align][align=center][img=,553,136]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537465105_7074_2222981_3.png!w553x136.jpg[/img][/align][align=center][/align][align=left][img=,579,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545307700_4011_2222981_3.png!w579x319.jpg[/img][/align][align=left][/align][align=left]为使客户有更多色谱柱选择,本实验室也尝试了能够在纯水系流动相下稳定使用的高极性色谱柱——CAPCELL PAK C[sub]18[/sub] AQ进行分析。如图3,几种头孢中间体的整体保留有所增强,而高浓度上样仍会出现与MGII色谱柱相似的过载现象;如图4,降低进样体积进行分析,可得到良好结果,同时发现扩环酸有一定程度的拖尾(见表2)。[/align][align=center][/align][align=center][img=,527,377]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546103944_1716_2222981_3.png!w527x377.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,525,374]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546123571_3070_2222981_3.png!w525x374.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表2 CAPCELL PAK C[sub]18 [/sub]AQ分析结果详表(进样量:5 μL)[/align][align=center][img=,558,135]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546125551_7090_2222981_3.png!w558x135.jpg[/img][/align][align=center][/align][align=left][img=,577,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101547424196_8227_2222981_3.png!w577x319.jpg[/img][/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 250 mm和高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 250 mm,以磷酸盐缓冲液(pH 6.0)-乙腈为流动相体系,在30°C柱温条件下进行梯度分析,均能够实现7-ADCA、△-2-7-ADCA、苯乙酸和扩环酸的良好分离,其中,CAPCELL PAK C[sub]18[/sub] MGII色谱柱所得峰形更佳。[/align]
我是岛津-2010,柱子为RTX-17MS,请问用什么条件能够将二者分开,另外,我的溶剂峰是很大的 啊
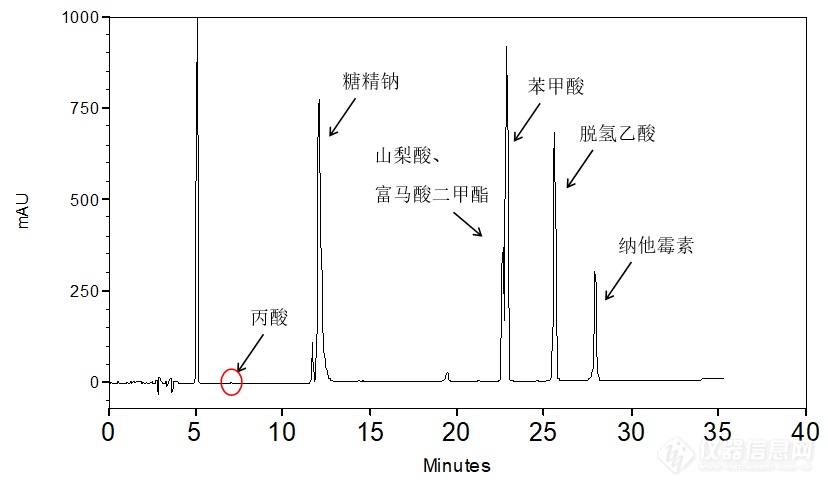
[align=center][b]食品中7种添加剂的共同分析——丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯[/b][/align][img=,99,45]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143105100_953_2222981_3.gif!w99x45.jpg[/img] [img=,121,46]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143267296_3413_2222981_3.gif!w121x46.jpg[/img] [img=,98,83]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143361658_9073_2222981_3.gif!w98x83.jpg[/img] [img=,89,50]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143485158_3959_2222981_3.gif!w89x50.jpg[/img] 丙酸钠 山梨酸 脱氢乙酸 苯甲酸[img=,112,150]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144042246_934_2222981_3.gif!w112x150.jpg[/img] [img=,270,166]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144144504_7297_2222981_3.gif!w270x166.jpg[/img] [img=,123,58]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144263678_2065_2222981_3.gif!w123x58.jpg[/img] 糖精钠 纳他霉素 富马酸二甲酯丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯为食品中常见的添加剂分析项目,客户希望通过一种方法来同时实现7种添加剂的分析,且整体分析时间要求尽量短,以提高工作效率。首先,使用大曹三耀(原资生堂)CAPCELL PAK系列色谱柱中的第一选择色谱柱——通用型反相柱[color=#3366ff][b]CAPCELL PAK C18 MGII[/b][/color]来进行方法初筛;考虑到7种添加剂中的酸性化合物较多,为取得良好保留,在流动相的选择方面,首先尝试在酸性条件下(0.1%磷酸)进行分析,结果如图1所示。在酸性条件下,苯甲酸、山梨酸和富马酸二甲酯三者间分离情况不佳。[align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104146524_289_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 酸性条件下分析结果[/align][img=,431,219]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104139552_9327_2222981_3.png!w431x219.jpg[/img]在此条件下,进一步对梯度条件进行调整,并将流动相中的乙腈更换为甲醇,更换不同种类色谱柱,但均未能获得良好结果。接下来我们将流动相pH改变,将0.1%磷酸溶液更换为20 mmol/L磷酸二氢铵溶液进行分析,结果如图2所示。能够看到7个较明显物质峰得到分离。[align=center][img=,690,359]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104574536_4474_2222981_3.png!w690x359.jpg[/img][/align][align=center]图2 磷酸二氢铵条件分析结果[/align][img=,450,220]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104576746_74_2222981_3.png!w450x220.jpg[/img]由于客户想要在短时间内得到良好分析,因此我们进一步将色谱柱由常规规格[b]S5 4.6 mm i.d. × 250 mm[/b]更换为[b][color=#3366ff]S3 4.6 mm i.d. × 100 mm[/color][/b],分析结果如图3所示。[align=center][img=,690,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105363445_4814_2222981_3.png!w690x418.jpg[/img][/align][align=center]图3 MGII小粒径短柱分析结果[/align][img=,459,221]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105376737_3055_2222981_3.png!w459x221.jpg[/img]但同时在分析中发现丙酸钠单标无法得到良好响应(图4),且富马酸二甲酯出峰行为异常(图5)。[align=center][img=,690,337]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106365666_7879_2222981_3.png!w690x337.jpg[/img][/align][align=center]图4 丙酸钠单标分析结果[/align][align=center] [/align]如图5结果所示,将7种添加剂各取100 μL后稀释到1 mL配制混合标准品时(第一次混合样品,终浓度为各0.1 mg/mL),未见富马酸二甲酯峰出现,此时再将此溶液与富马酸二甲酯单标以4:1比例混合后(第二次混合样品,富马酸二甲酯终浓度0.28 mg/mL,其他为0.08 mg/mL),富马酸二甲酯峰出现,同时2号峰峰面积增大。富马酸二甲酯单标进样无异常。因此富马酸二甲酯在混合溶液中可能存在[color=red]降解反应、溶解度不佳或其它原因[/color],导致出峰行为异常,建议对该物质性质进行考察,或进行方法学验证。[align=center][img=,690,450]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106370947_4629_2222981_3.png!w690x450.jpg[/img][/align][align=center]图5 富马酸二甲酯分析结果[/align][align=left] 综上所述,使用[b][color=#ff0000]CAPCELL PAK C[sub]18[/sub] MGII S3 4.6 mmi.d. × 100 mm[/color][/b]色谱柱可在[color=#3366ff][b]20 mmol/L磷酸二氢铵-乙腈[/b][/color]条件下基本实现7种常见食品添加剂的分析,但丙酸和富马酸二甲酯出峰异常,建议进一步进行方法学验证。[/align][align=left][/align][align=left] [/align][align=right] [/align][b][/b][align=right][b][color=#333333]LC Application Lab, Sanyofine China[/color][/b][/align][color=#333333][/color][align=right]Beijing, China[/align][color=#333333][/color][align=right]Phone 400 801 3103[/align]
如题,我分离的物质是褐藻胶中的甘露糖醛酸与古罗糖醛酸,此二者为葡萄糖的同分异构体,二者之间仅在C5位异构。水解后系列处理,用醋酸酐衍生为酯类,进行气相分析。所用的条件是SGE AC225,炉温215,检测器和进样口都是250.肌醇做内标。结果谱图相当混乱,有时出两个峰,有时出三个峰,而且谱图面积和出峰时间也不相同。请问如果要优化条件,该如何优化?柱温和检测器的温度么?优化的幅度是多大呢?相同物质的出峰时间因个人操作(手动进样)原因会有偏差,多大的偏差内是可以接受的呢?另外想查找下关于柱子的情况,始终查不到详细的资料。澳大利亚SGE公司的产品。本人是气相新手,求各位大侠指导!本想附谱图,但是太乱了,没有比较好的。
食品中重金属检测,有现场快速检测与实验室检测,除仪器的结构大小不一样外,二者方法的原理是一样的吗?
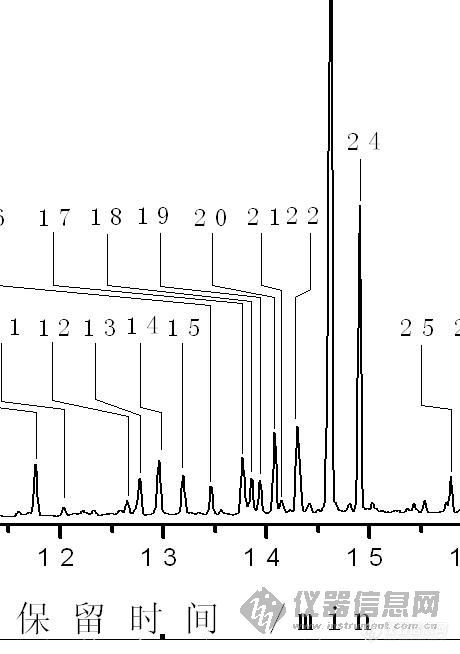
对含有松香的造纸白水硅烷化处理后进行GC-MS分析,GC图上有七个峰显示为松香的某种同分异构体,可是NIST05解谱显示这七个峰只代表三同分异构体衍生物,并且在该谱库中没有找到另外几同分异构体衍生物质谱图,质谱分析表明:下图中在17-25峰之间,除了第18、23峰外均为松香异构体的峰,怎么确定这七种物质的种类?请各位指点![em0910]E-mail: mykouer@163.com[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903071124_137172_1624214_3.jpg[/img]
我是做食品的,了解一些相关资料,如有需要我愿与您共同分享
最近安排做甲哌鎓和矮壮素的含量测定,发现用同一方法均可测出二者的含量,哪位有没有好的经验可以分享一下,可以将二者区分开来?







 我要推广仪器
我要推广仪器
 下载APP
下载APP