请问有没有办法用分光光度计测定二甲戊乐灵的残留浓度?
有没有谁分析过二甲戊灵原药跟二甲戊灵悬浮剂的啊,求分析方法!!!悬浮剂一般需要测哪些指标呢!谢谢啦
我们使用硫酸二甲酯作为原料,最有反应液用水进行稀释,稀释液位酸性,怎样检测酸性稀释液中的硫酸二甲酯残留?
1.分析目标化合物 二甲嘧菌胺 2.仪器设备 带碱热离子检测器或高灵敏度氮磷检测器的气相色谱仪和气相色谱—质谱仪。 3.试剂 丙酮 氯化钠溶液 正己烷 乙腈 无水硫酸钠 柱色谱用硅胶:将柱色谱用硅胶(粒径63~200μm)在130℃加热12小时以上,干燥器中放冷,加5%的水。 4.标准品 二甲嘧菌胺: 含二甲嘧菌胺99%以上,熔点为96℃~97℃。 5.试验溶液的制备 a 提取方法 ①豆类: 将样品粉碎,过420μm的标准网筛后,称取其10.0g,加入20mL水,放置2小时。 加入100mL丙酮,搅拌3分钟后,用涂布1cm厚硅藻土的滤纸抽滤于磨口减压浓缩器中。取出滤纸上的残留物,加入50mL丙酮,搅拌3分钟后,按上述同样操作,合并滤液于减压浓缩器中,40℃以下浓缩至约30mL。 将其移入预先注入100mL 10%氯化钠溶液的300 mL分液漏斗中。用100mL正己烷洗涤上述减压浓缩器的茄型瓶,合并洗液于上述分液漏斗中。用振荡器激烈振荡5分钟后,静置,正己烷层移入300mL三角瓶中。水层中加入50mL正己烷,按上述同样操作。合并正己烷层于上述300mL三角瓶中。加入适量无水硫酸钠,不时振荡、混合,放置15分钟后,滤入磨口减压浓缩器中。再用20mL正己烷洗涤三角瓶,以此洗涤液洗涤滤纸上的残留物,重复操作二次。合并两洗涤液于减压浓缩器中,在40℃以下除去正己烷。 残留物中加入30mL正己烷,移入100mL分液漏斗中。加入30mL正己烷饱和乙腈,用振荡器激烈振荡5分钟后,静置,乙腈层移入磨口减压浓缩器中。正己烷层中加入30mL正己烷饱和乙腈,按上述同样操作,重复操作两次。合并乙腈层于减压浓缩器中,40℃以下除去乙腈。残留物中加入5mL正己烷溶解。
有谁用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]研究过二甲戊灵、双甲脒、百菌清、甲霜灵这四种农药的残留阿?有的话,用的什么前处理,效果如何?
食品中富马酸二甲酯残留量的测定 1 范围 本方法规定了食品中富马酸二甲酯残留量的GC测定方法。 本方法适用于粮食、糕点、水果等食品中富马酸二甲酯残留量的测定。 本方法的检测限(LOD)为:25mg/kg,最低检出浓度为25ug/ml。 2 规范性引用文件 下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。 GB/T 6682 分析实验室用水规格和试验方法 3 原理 样品中富马酸二甲酯(DMF)经提取净化后,用附氢火焰离子检测器的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]进行分离测定,与标准系列比较定量 4 试剂和材料 4.1除非另有说明,所有试剂均为分析纯。水为符合GB/T 6682规定的一级水 4.2氯仿 4.3无水硫酸钠。 4.4中性氧化铝(层析用60-80目)。 4.5标准溶液贮备液:0.1g富马酸二甲酯(含量99.9%),用少量氯仿溶解,转移到100ml容量瓶中,用氯仿稀释至刻度,该标准溶液含富马酸二甲酯1mg/ml。 4.6标准溶液使用液:分别吸取标准溶液5、10、15、20、25、30ml于100ml容量瓶中,用氯仿稀释至刻度,富马酸二甲酯浓度分别为50、100、150、200、250、300ug/ml。 5 仪器与设备 5.1[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],附氢火焰离子检测器 5.2匀浆机。 5.3粉碎机。 6 分析步骤 6.1样品制备 6.1.1 粮食、糕点、及含水分少低脂类的固体食品 称取5.0g或10.0g粉碎样品,置于250ml具塞三角烧瓶中,加30ml氯仿,振摇30min,用定性滤纸过滤,取10ml滤液,吹入氮气使浓缩至1ml,备用。 6.1.2 含脂肪较多的样品 称取粉碎样品10.0g,加中性氧化铝5-10g(视脂肪多少而定),以下按6.1.1“加30ml氯仿…”起,依法操作。 6.1.3 水果类 将水果去皮,切成碎片,加等量蒸馏水于匀浆机中匀浆后,称取20.0g匀浆液(相当于10g样品),加氯仿30ml,振摇30min,用定性滤纸过滤于125ml分液漏斗中,待分层后,用无水硫酸钠过滤,取滤液10ml,吹入氮气浓缩至1ml,待测。 6.2 测定 6.2.1色谱参考条件 6.2.1.1色谱柱: 玻璃柱(内径3mm,长2m),内装涂以2%OV-101和6%OV-210混合固定液的60-80目Chromosorb W.AW DMCS(HP) 6.2.1.2气流速度:氮气50ml/min 空气500ml/min 氢气35ml/min 。 6.2.1.3温度:气化室及检测器200℃,柱温155℃。. 6.2.1.4进样量:1μL。 6.2.2 测定 注入1uL标准系列中各浓度标准使用液于[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]中,测得不同浓度富马酸二甲酯的峰高,以浓度为横坐标,相应的峰高值为纵坐标,绘制标准曲线。同时注射一定体积样品溶液,测得峰高与标准曲线比较定量。 6.2.3 阳性样品的确证 按照上述条件测定试样和标准工作溶液,如果试样中的质量色谱峰保留时间与标准工作溶液一致(变化范围在±2.5%之内) 条件许可可以通过GC—MS定性 6.2.4 空白实验 除不称取样品外,均按上述测定条件和步骤进行。 6.2.5 允许差 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。 7. 结果计算 样品中富马酸二甲酯残留量按照下式计算:[align=center][img]http://img.vogel.com.cn/2013/0426/0858274456.jpg[/img][/align] 7. 1相关技术参数 方法最低检出限:25mg/kg。回收率在88.9%~94.2%范围内,其相对标准偏差在4.32%~9.07%的范围内。
检测甲苯 和二甲亚砜溶剂残留,但是样品沸点在220度,结果直接进样样品有峰和甲苯重合,我用甲醇作溶剂有溶剂峰,样品品峰很大,怎么办啊?我的色谱条件:DB-624 初始40度5min,20度升温至200度
苯菌灵遇有机溶剂或水会分解成多菌灵和异氰酸丁酯,在液相分析中,也会发生柱上分离,使得两峰不能基线分离,所以苯菌灵的残留分析,我感到很困难,有老师做过这方面的工作吗,能给些建议吗[em46]
附件2食品中富马酸二甲酯残留量的测定 ([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法)1 范围 本方法规定了食品中富马酸二甲酯残留量的GC测定方法。本方法适用于粮食、糕点、水果等食品中富马酸二甲酯残留量的测定。本方法的检测限(LOD)为:25mg/kg,最低检出浓度为25ug/ml。2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法3 原理样品中富马酸二甲酯(DMF)经提取净化后,用附氢火焰离子检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进行分离测定,与标准系列比较定量4 试剂和材料4.1除非另有说明,所有试剂均为分析纯。水为符合GB/T 6682规定的一级水4.2氯仿4.3无水硫酸钠。4.4中性氧化铝(层析用60-80目)。4.5标准溶液贮备液:0.1g富马酸二甲酯(含量99.9%),用少量氯仿溶解,转移到100ml容量瓶中,用氯仿稀释至刻度,该标准溶液含富马酸二甲酯1mg/ml。4.6标准溶液使用液:分别吸取标准溶液5、10、15、20、25、30ml于100ml容量瓶中,用氯仿稀释至刻度,富马酸二甲酯浓度分别为50、100、150、200、250、300ug/ml。5 仪器与设备5.1[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],附氢火焰离子检测器5.2匀浆机。5.3粉碎机。6 分析步骤6.1样品制备6.1.1 粮食、糕点、及含水分少低脂类的固体食品 称取5.0g或10.0g粉碎样品,置于250ml具塞三角烧瓶中,加30ml氯仿,振摇30min,用定性滤纸过滤,取10ml滤液,吹入氮气使浓缩至1ml,备用。6.1.2 含脂肪较多的样品 称取粉碎样品10.0g,加中性氧化铝5-10g(视脂肪多少而定),以下按6.1.1“加30ml氯仿…”起,依法操作。6.1.3 水果类 将水果去皮,切成碎片,加等量蒸馏水于匀浆机中匀浆后,称取20.0g匀浆液(相当于10g样品),加氯仿30ml,振摇30min,用定性滤纸过滤于125ml分液漏斗中,待分层后,用无水硫酸钠过滤,取滤液10ml,吹入氮气浓缩至1ml,待测。6.2 测定6.2.1色谱参考条件6.2.1.1色谱柱: 玻璃柱(内径3mm,长2m),内装涂以2%OV-101和6%OV-210混合固定液的60-80目Chromosorb W.AW DMCS(HP) 6.2.1.2气流速度:氮气50ml/min;空气500ml/min 氢气35ml/min;。6.2.1.3温度:气化室及检测器200℃,柱温155℃。.6.2.1.4进样量:1μL。6.2.2 测定注入1uL标准系列中各浓度标准使用液于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]中,测得不同浓度富马酸二甲酯的峰高,以浓度为横坐标,相应的峰高值为纵坐标,绘制标准曲线。同时注射一定体积样品溶液,测得峰高与标准曲线比较定量。6.2.3 阳性样品的确证按照上述条件测定试样和标准工作溶液,如果试样中的质量色谱峰保留时间与标准工作溶液一致(变化范围在±2.5%之内)条件许可可以通过GC—MS定性6.2.4 空白实验除不称取样品外,均按上述测定条件和步骤进行。6.2.5 允许差在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。7. 结果计算样品中富马酸二甲酯残留量按照下式计算: X:样品中富马酸二甲酯残留量,mg/kgA:测定样品液中富马酸二甲酯含量,ug/mlV1:浓缩用样品提取液体积,mlV2:样品氯仿提取液总体积,mlV3:样品浓缩后的体积,mlV4:标准溶液进样体积,ulV5:样品溶液进样体积,ulm:样品重量,g7. 相关技术参数方法最低检出限:25mg/kg。回收率在88.9%~94.2%范围内,其相对标准偏差在4.32%~9.07%的范围内。
我正在做药品中二甲胺的残留分析,有哪位老师用常规气相方法检测过?谢谢
[em06] 各位高手,我想问一下有没有人知道恶霉灵的农药残留分析方法,有做过的高手请帮帮我了。我用的甲醇做溶剂,流动相是甲醇+水=7+3,波长:212,流速:1单恶霉灵出峰时间在2.9min,而溶剂在这里也有一个大峰,分不开。很苦闷,求各位有什么好的方法和文献,指教一下!!谢谢大家了!!![em24]
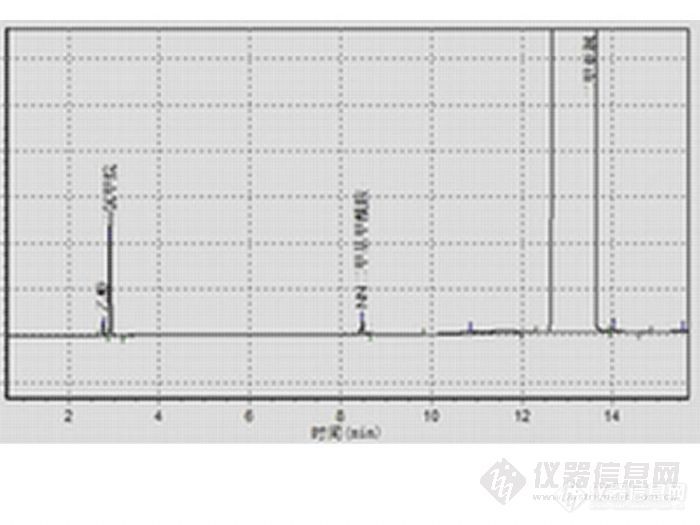
顶空毛细管柱气相色谱法分析测定药品中残留溶剂http://ng1.17img.cn/bbsfiles/images/2011/05/201105211013_295254_2242538_3.jpg 摘要 目前国家对药品中残留溶剂的检测还没有一个统一的国家标准,在关于有机溶剂残留量的指导原则中将二氯甲烷、DMF列为第二类必须控制的毒性试剂,将乙醇、丙酮列为第三类低毒性的溶剂。建立一种快速测定药品中残留溶剂的分析方法很有必要。为此南京科捷采用DK-300A顶空进样器取样,气相色谱毛细管柱分离,分析测定药品中常见的残留溶剂(乙醇、二氯甲烷、NN二甲基甲酰胺、二甲亚枫)。结合相关指标,本实验将二氯甲烷的限量规定为 600ppm,DMF的限量规定为 880ppm,乙醇及丙酮的限量规定为 5000ppm。实验结果表明:顶空毛细管柱气相色谱法快速、简便、准确是测定药品中残留溶剂较为理想的方法。关键词 乙醇 二氯甲烷 NN二甲基甲酰胺 顶空进样 毛细管气相色谱 药品 有机溶剂残留1.药品中乙醇、二氯甲烷、NN二甲基甲酰胺色谱图峰序1. 乙醇2.二氯甲烷3.NN二甲基甲酰胺4.二甲亚枫(溶剂)2.本方法应用范围在合成原料药,辅料或制剂生产的过程中使用或产生的挥发性有机化学物质,它们在实际的生产中未能被完全地清除。本方法可应用在药品中残留溶剂的检测中。近年来,药品中残留有机溶剂的毒性和致癌作用日益引起各方面的重视。药品中残留有机溶剂于1997年被美国FDA列为药品监控项目。我国药品中残留有机溶剂检测也越来越受到有关方面的重视。本文初步研究探索采用顶空毛细管柱气相色谱法分析药品中残留挥发性有机溶剂。顶空气相色谱法只将挥发和半挥发的组份引入柱子,可避免非挥发性的物质对系统的污染,样品前处理简便,分析效率高。结果表明,本方法快速、准确、重现性好。3.仪器及试剂配置色谱仪器配置色谱柱及试剂GC5890(FID检测器)毛细管专用柱30*0.32.*0.5乙醇、二氯甲烷各一瓶顶空进样器:DK-300ANN二甲基甲酰胺1瓶N2000色谱工作站(电脑自备1台)二甲亚枫1瓶氢氮氧一体发生器或钢瓶气各一瓶顶空压盖机1台顶空瓶20ml (带塞) 50只
求助各位专家啦,我们用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]分析农药残留怎么总会有邻苯二甲酸类的干扰物啊?这个是从哪里带入的呀?[em09512]
编者按:药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。除上述这三类溶剂外,在药物、辅料和药品生产过程中还常用其他溶剂,如1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙酮、甲基四氢呋喃、石油醚、三氯乙酸、三氟乙酸。这些溶剂尚无基于每日允许剂量的毒理学资料,如需在生产中使用这些溶剂,必须证明其合理性。美、日、欧洲的药典对这一问题的处理不一样,被列入清单的毒性有机溶剂的种类和相应的可接受限度也不相同。虽然国际协调大会(ICH)关于药品中残留溶剂的指导原则在1997年就已生效,但《美国药典》至今尚未完全采纳该指导原则。《美国药典》的残留溶剂检测归在附录中的“有机挥发性杂质”篇,规定只有在生产商指出产品中可能有残留溶剂存在时才进行此检测,而当生产商根据其产品的生产、运输和储藏的相关知识可以保证产品中无某一种溶剂存在,并且保证如果进行此检测的话,产品能符合残留限度要求的时候,就可不进行此检测。同时还认为,装在气密性容器中的物品在运输过程中不受任何溶剂的污染。《美国药典》推荐进行苯、氯仿、二氧杂环己烷、亚甲基氯、三氯乙烯残留量检测。此外,还在一些药品的各论中指定进行环氧乙烷的残留量检测,除非另有规定,环氧乙烷残留量的可接受限度为10ppm。除此以外,《美国药典》不考虑国际协调大会(ICH)指导原则中的其他溶剂。第14版《日本药典》已采用国际协调大会(ICH)的指导原则,将残留溶剂定义为存在于药品中,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行检测,限度符合国际协调大会(ICH)指导原则规定的有机溶剂。《欧洲药典》完全采纳国际协调大会(ICH)关于残留溶剂的指导原则。第4版《欧洲药典》叙述了如何对第一类和第二类溶剂进行鉴别和定量分析的方法,试验方法还适用于第三类溶剂和限度大于1000ppm(0.1%)的第二类溶剂的定量分析。
[color=#444444]求教DMSO(二甲基亚砜)残留量检测方法 ,本人正在做处理二甲基亚砜废水的实验,但是不知道怎么检测其中反应后残留的二甲基亚砜的含量,实验室有SP-6890型的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],请问能用这个检测吗,如果能又该怎么设置升温程序呢,需要加内标物吗?[/color]
做药物的有机溶剂残留,用二甲基亚砜做溶解药品的溶剂,二甲基亚砜本身有好多小杂质峰干扰药物中的待测有机溶剂的测定,有谁用过二甲基亚砜做过溶解样品的溶剂的,在2-5min是不是也有好多小杂质峰干扰测定?换了好几个厂家的二甲基亚砜了,或多或少都有干扰,如何处理?还有什么其它溶剂可用没有,甲醇、甲苯、正丁醇、正己烷做为溶剂药物都不溶解(我测药物中乙醇,乙酸乙酯、二氯甲烷、四氢呋喃、NN-二甲基甲酰胺的残留)
整理了一些用气象色谱分析的应用资料,供大家分享! 今后会陆续整理上传应用资料的,请大家关注哦!1. 10类农药残留的分析方法2. 氨纶生产过程中胺的分析3. 包装食品中溶剂残留的分析4. 分析薄膜包衣片中的二氯甲烷5. 顶空[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析护肤品中芳香成分(甲基吡嗪)6. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析溶剂纯度(氯仿)7. 挥发性有机酸的分析(氯乙酸,[font=Arial]三氯乙酸,[font=MingLiU]溴乙酸,[font=MingLiU]二氯乙酸,二溴乙酸)8. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析饮用水中残留农药成分9. 牙膏增白剂中有毒气体的分析 10. 液相色谱法分析乳酸钠11. 在线分析工业中痕量废气12. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析烟草中苯、异丁酯、正丁醇、丙二醇甲醚等挥发性有机物13. 永久性气体分析系统分析标准气样的方法14. 对变压油中气体的分析15. 热裂解[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法对聚乙烯的分析16. 热裂解[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法对尼龙66的分析17. 食用色素中残留溶剂的分析方法18. 氨基酸分析中的两种衍生化方法19. 食用包装材料中残留溶剂的分析方法20. 一般残留溶剂的分析方法 (丙酮,四氢呋喃,乙酸乙酯,甲醇,二氯甲烷,苯,乙腈,氯仿,甲苯)21. 药物中残留溶剂的分析方法2[/font][/font][/font][font=MingLiU] (二氯甲烷[/font][font=宋体],[/font][font=MingLiU]氯仿[/font][font=宋体],[/font][font=MingLiU]三氯乙烯[/font][font=宋体],[/font][font=MingLiU]1,4-[/font][font=MingLiU]二氧六环[/font])22. 药物中残留溶剂的分析方法[font=MingLiU] (甲醇[/font][size=3][font=Times New Roman] [/font][/size][font=Times New Roman][/font][font=宋体],[/font][font=MingLiU]丙酮[/font][font=宋体],[/font][font=MingLiU]乙酸乙酯[/font][font=宋体],[/font][font=MingLiU]1.4-[/font][font=MingLiU]二氧六环[/font][font=宋体],[/font][font=MingLiU]吡啶[/font][font=宋体],[/font][font=MingLiU]二甲基甲酰胺[/font][color=black][font=MingLiU])23. 应用PDECD检测器分析农药类物质24. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测试酒样中31种混标25. 顶空法检测制糖原材料中溶剂残留(甲醇、乙醇)[/font][/color]
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。第二类溶剂是指无基因毒性但有动物致癌性的溶剂。按每日用药10克计算的每日允许接触量如下,乙腈(410ppm)、氯苯(360ppm)、氯仿(60ppm)、环己烷(3880ppm)、二氯甲烷(600ppm)、二氧杂环己烷(380ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、2-乙氧基乙醇(160ppm)、2-甲氧基乙醇(50ppm)、环丁砜(160ppm)、1,2,3,4-四氢化萘(100ppm)、嘧啶(200ppm)、甲苯(890ppm)、甲酰胺(220ppm)、1,2-二氯乙烯(1870ppm)、N,N-二甲基乙酰胺(1090ppm)、N,N-二甲基甲酰胺(880ppm)、乙烯基乙二醇(620ppm)、正己烷(290ppm)、甲醇(3000ppm)、甲基环己烷(1180ppm)、N-甲基吡咯烷酮(4840ppm)、二甲苯(2170ppm)。第三类溶剂是指对人体低毒的溶剂。急性或短期研究显示,这些溶剂毒性较低,基因毒性研究结果呈阴性,但尚无这些溶剂的长期毒性或致癌性的数据。在无需论证的情况下,残留溶剂的量不高于0.5%是可接受的,但高于此值则须证明其合理性。这类溶剂包括戊烷、甲酸、乙酸、乙醚、丙酮、苯甲醚、1-丙醇、2-丙醇、1-丁醇、2-丁醇、戊醇、乙酸丁酯、三丁甲基乙醚、乙酸异丙酯、甲乙酮、二甲亚砜、异丙基苯、乙酸乙酯、甲酸乙酯、乙酸异丁酯、乙酸甲酯、3-甲基-1-丁醇、甲基异丁酮、2-甲基-1-丙醇、乙酸丙酯。
各位朋友,请教个问起:用二甲基亚砜作溶媒精制,样品要检测二甲基亚砜(ICH中的3类溶媒)的残留量,请问要选择什么溶剂来溶解样品?选用什么色谱柱合适?
二甲苯溶剂残留怎么计算??各个异构体比例没有标明,只标有乙苯19%;计算用峰面积和,再按外标法计算吗?如果是的话,也包括乙苯吗???
2012年8月1日,美国联邦法典CFR TITLE 21§180.518部分修订对农药二甲嘧菌胺Pyrimethanil的残留限量要求。 删除对下列物质的残留限量要求:产品中文名称产品英文名称限量要求葡萄Grape5 ppm洋葱鳞茎Onion, bulb0.1 ppm青洋葱Onion, green2 ppm草莓Strawberry3 ppm 新增对下列物质的残留限量要求:产品中文名称产品英文名称限量要求缓生浆果,第13-07G亚组Berry, low growing, subgroup 13–07G3 ppm小型蔓生水果,13-07F亚组,除猕猴桃Fruit, small, vine climbing, subgroup 13–07F, except fuzzy kiwifruit5 ppm人参Ginseng1.5 ppm洋葱,鳞茎,第3-07A亚组Onion, bulb, subgroup 3–07A2 ppm青洋葱,第3-07B亚组Onion, green, subgroup 3–07B3 ppm 美国农药残留限量要求可登录下述网址查询:http://www.tbt-sps.gov.cn/FOODSAFE/XLBZ/Pages/pesticide
近日在某省公安厅做火灾爆炸现场助燃剂残留物分析(常见的助燃剂汽油、柴油、航空煤油和油漆稀释剂等),主要分析里面的支链烷烃、苯系物(主要是甲苯、乙苯、二甲苯、三甲苯、四甲苯等);与其分析人员进行了相关的经验交流得知,老的方法都是从现场取一些物证如布料、土壤后拿回来采用溶剂洗脱提取,但是往往由于含量较低反而在这个洗脱提取的过程中带入了新的干扰影响分析,一直找不到比较好的处理方法,也试着用传统顶空做但效果不是很好,因在燃烧过程中低沸点的如苯、甲苯、乙苯等都已燃烧完全,残留量很小很小,相反里面的高沸点的物质如三甲苯、四甲苯等而不易燃烧充分,残留量较大,传统顶空做高沸点组分不是很理想。 该用户有一套大体积顶空,加热温度能达到300度左右,于是我们就想着用该套设备来试试,把样品直接放入到大体积顶空,然后温度设定到200度用氦气不断吹过,另一端接入到装有三合一填料的捕集阱进行富集,富集一段时间后然后解析这根捕集阱进样分析,经过多次试验取得了很好的效果,采用这套装置后简化了样品前处理过程,但是在分析过程中还是有新的问题发生,就是大体积顶空还是容易残留小部分样品,但是后来在共同努力下解决了这些小问题,因为整个过程中我一直参与其中,故想把这些东西记下来与大家共同探讨下,我觉得把整个流程记录下来最终总结应该是一个好的论文题材。 接下来的时间我来介绍下什么是大体积顶空以及其工作原理,有机会我会发一些图片上来与大家一起交流,大体积顶空是介于静态顶空与吹扫捕集间的一种样品前处理设备(个人见解),所谓大体积,就是容纳样品的罐子较大,我这次使用过的大约有电饭煲那么大小,所有内壁及管线均是采用惰性脱活的硅烷化处理的,这个罐子的加热温度能到400度左右,这些特点决定了可以处理的样品量大,可处理的样品沸点较高,燃烧后样品残留较小。样品导入捕集阱有两种模式,一种是常见的吹扫模式即通入一路吹扫气将燃烧后的气体带入,另外一种方法则是采用泵反抽的模式,采用泵抽的模式可以降低吹扫气带入干扰的风险,吸附阱的填料tenax TA & GR,活性炭三合一的,增加对各物质的吸附效果。(出差中,先记下这些,未完待续)
请提供一下蔬菜和土壤中甲基硫菌灵的农药残留量分析方法!谢谢~!
求助甲磺隆的残留分析方法,手上有US EPA残留报告,但是感觉不是很好
[font=宋体][b]1.前言[/b]邻苯二甲酸酯类增塑剂是一类增加聚合物树脂的可塑性、增强制品柔软性的助剂,也是迄今为止产量和消费量最大的助剂种类,其中邻苯二甲酸酯类的使用最为广泛。邻苯二甲酸盐由于未聚合到塑料基质中,随着使用时间的推移,可由塑料中转移到环境中,造成污染。也可通过呼吸、饮食和皮肤接触直接进入人体,导致肝肾功能下降,具有致突变性、致癌性。全球已有很多国家通过立法限制使用邻苯二甲酸酯类增塑剂,包括欧盟 REACH 法规和美国的《消费品安全改进法》等。增塑剂在食品安全引起关注,首先是 2011 年 5 月起台湾食品中先后检出 DEHP、DINP、DNOP、DBP、DMP、DEP 等 6 种邻苯二甲酸酯类塑化剂成分,药品中检出DIDP。截至6月8日,台湾被检测出含塑化剂食品已达 961 项。6 月 1 日卫生部紧急发布公告,将邻苯二甲酸酯(也叫酞酸酯)类物质,列入食品中可能违法添加的非食用物质和易滥用的食品添加剂名单。近段时间,白酒中检测出塑化剂的风波再次引起大家的关注,白酒添加塑化剂则有可能是为了让年份不够的酒液看起来好看,增加各种增粘剂可固化伪造粮食酒内的糖分,产生粘杯挂杯的效果。但同时也有可能是白酒产品中的塑化剂属于特定迁移,主要是生产或包装过程中与塑料制品接触,塑化剂被酒精溶出所造成的。目前国内颁布了 [url=https://www.antpedia.com/standard/5741037.html]GB/T 21911-2008[/url]《[url=https://www.antpedia.com/standard/5741037.html]食品中邻苯二甲酸酯的测定[/url]》的检测方法。本实验在此标准基础上,进行优化,采用水浴加热去除乙醇后,正己烷提取,采用赛默飞世尔科技全新一代三重四极杆[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱联用仪(TSQ 8000)分析检测白酒中 16 种邻苯二甲酸酯的方法。通过二级质谱扫描充分减少了在复杂基质样品中的背景干扰影响,提高了目标化合物的检测灵敏度。[b]2.实验部分[/b]2.1 仪器和试剂 质谱仪 : TSQ 8000 质谱仪 ( 赛默飞世尔科技,美国 ); [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]:Trace1310[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff] GC [/color][/url]配 AI l310 自动进样器 ( 赛默飞世尔科技,美国 ); 色谱柱 : TR-PesticideII 30 m* 0.25 mm* 0.25 μm 毛细管色谱柱(带 5 m 预柱); 试剂:正己烷,农残级; 白酒:自购于超市。2.2 仪器方法[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]方法: 柱温箱:60oC 保持 1 min,以 20oC/min 升至 220oC,保持1 min,再以 5oC/min 的速率升至 280oC,保 持 3 min; 进样口:不分流进样,不分流时间:1 min,衬管:惰性不分流(货号:453A1925),进样口温度为 250oC;载气:恒流,1 ml/min; 传输线:280oC质谱方法:离子源温度为 250 oC,采用 Acquisition-Timed 方法,SRM扫描,具体检测离子对如表 1 所示:[b]3. 实验结果分析[/b]3.1 全自动二级质谱条件的优化(Auto-SRM)在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-三重串联四极杆质谱联用检测过程中,为了保证定性和定量的准确,必须对待测物的离子对(母离子和子离子)、碰撞能量、扫描时间、驻留时间及监测反应离子的数目等一系列质谱参数进行优化,以期达到最佳的灵敏度。使用TSQ 8000 [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url],可采用特有的 Auto-SRM 模式,可全自动完成所有化合物的二级质谱参数优化,并自动给出碰撞能量优化结果,简单直观,如图 1 所示,大大节省了分析时间。本实验在固定色谱条件下,通过优化后的仪器参数,样品色谱图与标准物质色谱图比较,保留时间相对偏差不超过标准物质±2.5%;采用多离子对定性,相对离子丰度最大允许偏差不超过 ±20%,确保了定性的准确度。3.2 色谱分离结果由于溶剂中可能含有邻苯二甲酸酯类物质,所以每次做样前我们都做了溶剂对照,以便排除溶剂中的干扰效应,摒除假阳性结果。由于 16 种邻苯二甲酸酯的离子对信息有一些比较相似,所以我们选择了分离度比较好的弱极性色谱柱,从而保证了 16种邻苯二甲酸酯在色谱上得以分离,使定性定量更加准确。16种邻苯二甲酸酯色谱分离情况如图 2 所示:[/font][align=center][img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202537114958.png[/img][/align][align=center][img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202545120917.png[/img][img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202552172975.png[/img][/align][font=宋体][b]3.3 标准曲线及最低定量限[/b]以正己烷为溶剂,分别配制 1 μg/L,5 μg/L,10 μg/L,50 μg/L,100 μg/L,500 μg/L 的混标溶液,建立标准曲线,各化合物的标准曲线如表 2 所示,相关系数 R 2 均大于 0.991,表明这16 种化合物的标准曲线线性良好。由于溶剂本底中部分邻苯二甲酸酯是存在干扰的。所以需要将标准溶液扣除本底后再计算标准曲线,以使标准曲线更加准确。本次实验中 16 种邻苯二钾酸酯类化合物的最低定量限均为 1 μg/L,见图 3。其中溶剂中的 DEP、DIBP、DBP 和 DEHP 含量估计是 1 μg/L 标准品的20-30%,所以最低定量限设定为 1 μg/L。[/font][img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202570777932.png[/img][img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202582428175.png[/img][font=宋体][b]3.4 方法精密度和回收率的测定[/b]称取白酒样品,分别添加 2 个水平浓度的标准品,选取其中低浓度水平平行测定 6 次。结果表明,平均回收率为 81.5- 115%,相对标准偏差(RSD, n=6)为 1.74-4.95%。回收率和精密度数据结果见表 3。表3: 方法的精密度和回收率[img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202589628411.png[/img][/font][font=宋体][b]3.5 实际样品检测[/b]按照上述前处理方法,对市售的 4 种不同的白酒进行邻苯二钾酸酯类残留分析检测。测试结果统计见表 4。其中超过线性范围的采用二次稀释后定量。表 4:实际样品检测结果统计[img=image.png]https://i4.antpedia.com/attachments/att/image/20181126/1543202598516277.png[/img][/font][font=宋体]对检出的化合物可以通过二级质谱定性离子与定量离子的离子比例进一步确证化合物,下图显示了样品 1 中检出的 DIBP 的离子比例符合标准值,因此可以确证样品 1 中确实检出的是DIBP。[/font][font=宋体]根据 GB9685-2008《[url=https://www.antpedia.com/standard/5803603.html]食品容器、包装材料用添加剂使用卫生标准[/url]》的规定,邻苯二甲酸酯类物质是可用于食品包装材料的增塑剂,不是食品原料,也不是食品添加剂,严禁在食品、食品添加剂中人为添加。食品、食品添加剂中的邻苯二甲酸二 (2- 乙基己 ) 酯 (DEHP) 和邻苯二甲酸二正丁酯 (DBP) 最大残留量分别为 1.5 mg/kg 和 0.3 mg/kg。我们的样品测定结果表明, DIBP、DBP、DEHP 在白酒中普遍存在,其中1个样品中DBP,DEHP的含量超过规定,样品4中 DBP含量达2.885mg/L即样品含量为1.154 mg/kg,超标285%;DEHP的含量高达11.606 mg/L 即样品含量为4.64 mg/kg,超标 209%。[/font][font=宋体][b]4.结论[/b]本方法采用 ThermoFisher 公司全新一代三重四极杆质谱 TSQ 8000 测定白酒中 16 种邻苯二甲酸酯类物质残留,此方法具有操作方便,选择性好,线性范围宽,高灵敏度等特点,高灵敏度能够减少取样量,极大降低样品对质谱仪器的污染,节约了分析和维护成本。同时 TSQ 8000 提供的离子对扫描可以有效排除假阳性的干扰,使检测结果更加准确。在 100、20 μg/L 两个添加水平下,回收率为 81.5-115%,20 μg/L 浓度水平下相对标准偏差(RSD, n=6)为 1.74-4.95%。该方法最低定量限为1 μg/L,完全可以满足白酒中邻苯二甲酸酯类物质的检测需要[/font]
整理了几份分析方法,供大家参考!1. 分析对苯二酚葡萄糖苷2. 分析药物中的残留溶剂3. 5类有机磷农药化合物的分析方法
我们遇到一个困难,就是要检测固体药品中是否残留硫酸二甲酯,并作验证,要求检测限很低,我们试了很多方法均不行,GC主要是检测限太高,GC-MS主要是找不到一种合适的溶剂,硫酸二甲酯在水中易分解,所以只能选择有机溶剂,但我们的固体样品是个盐酸盐,在有机溶剂中溶解度不是很好,试过甲醇,硫酸二甲酯在甲醇中易转化且样品有严重干扰,所以不行,DMF 和DMSO也试过,我们的样品在DMF中根本不溶,在DMSO中要好一些,但是DMSO又跟硫酸二甲酯峰会重叠,也不行。不知道哪位高人有做过这方面的研究,或者经验丰富的给一点建议
杀菌剂多残留分析方法 检测农产品中农药残留量是评价其是否超过MRL值的前提,因此农药残留分析方法得到各国的重视。无论是多残留分析(MRMs)还是单残留分析(SRMs),均基于相似的检测步骤,多残留分析由于可同时检测多种农药残留的存在,因此通常是首选方法。公职分析化学家协会(AOAC)的方法是国际公认的多残留分析方法,可用于多种农药残留的检测。该方法通常用水溶性的溶剂提取,紧接着的净化用不溶于水的溶剂进行分配,再用硅胶或弗罗里硅土净化,净化后的提取物用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、高效液相色谱检测。尽管AOAC方法可以检测多达325种农药及相关化合物,但也存在一些不足之处,如效率低,不适于进行筛选分析;有些试剂有毒且用量大;不能分析一些新农药等。针对上述缺陷,一些新的提取、净化方法得到重视和发展,如固相萃取(solid-phase extraction,SPE)、固相净化(solid-phase cleanup,SPC)、基质固相分散萃取(matrix solid-phase dispersion,MSPD)、超临界流体萃取(supercritical fluid extraction,SFE)等。这些技术的突出特点是简便、样量小型化、萃取的广泛性。检测技术上,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]及高效液相色谱仍然是主要的技术手段。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方面,采用微波诱导等离子(microwave induced plasma,MIP)-原子发射检测器(atomic emission detector,AED)及多级质谱的检测技术取得了快速的发展;离子及离子对色谱以及柱后衍生技术的应用是高效液相色谱检测的研究热点,激光诱导荧光检测器也已开始应用。超临界流体色谱(Supercritical fluid chromatography, SFC)在农药残留检测中的成功应用,实现了样品中农药残留的提取、净化、检测一步完成,是当前联用技术的典型代表。2.1 提取从样品中提取残留农药的效果,很大程度上决定于农药的极性及样品基质的类型。提取过程通常将样品置于匀浆瓶中,添加溶剂,利用匀浆器(homogeniser)、搅拌机(blender)或超声波发生器(sonicator)匀浆。常用的有机溶剂有丙酮、乙腈、甲醇、乙酸乙脂等,根据样品类型、含水量及目标农药,有时需要添加适量的水或调整pH值。多数情况下样品能均匀的分散在有机溶剂中,从而可提高被测残留物的回收率,减少共提的干扰物比率。经均化作用后,以过滤或离心的方法将溶剂和固体分开。2.1.1乙腈提取法乙腈提取法可应用于大多数农药和其它一些化合物的提取,然而在该方法中,许多水溶性(极性)化合物在石油醚从乙腈水中提取农药以及在弗罗里硅土柱层析或氧化镁/硅藻土柱进行层析净化时,水溶性化合物或部分或全部损失。尽管如此这种提取方法仍适用于许多杀菌剂的分析。在Liao等人的方法中采用乙腈进行提取,通过添加氯化钠使乙腈与水分离,上层部分(乙腈)浓缩至小体积后用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]检测。AOAC早期的方法及美国加州食品和农业部 (California Department of Food and Agriculture,CDFA)的MRSM (Multiresidue Screen Method) 方法多采用乙腈作为溶剂。2.1.2丙酮提取法由于丙酮具有无毒、易于纯化、与乙腈和其它一些溶剂相比挥发性好而且价格低廉等优点,因此也是一种常用的提取溶剂,许多方法采用它。此外,在样品中含糖时,丙酮不会象乙腈那样与水形成两相,故可用于高含糖量的样品。实验表明,丙酮具有广泛的化合物和样品基质适用性,已有的回收率数据包括400余种化合物,其中含杀菌剂40余种。理论上,丙酮可用于提取任何样品中除了带有永久离子之外的残留农药。因此,许多国家农药残留标准方法均采用丙酮作为主要溶剂,如德国的DFG S19方法,美国FDA MRMs方法等。在这些方法中,丙酮提取液用氯化钠或硫酸钠饱和之后,分配至二氯甲烷、己烷或石油醚中,从而可得到对不同化合物有利的分配特性和有机相的快速分离。2.1.3乙酸乙酯提取法乙酸乙酯极性相对丙酮、乙腈要弱,因此其对弱极性农药的提取回收率一般较好些,并且其共提物尤其是色素要显著少于丙酮,从而减少了净化时的压力。采用乙酸乙酯作提取溶剂的方法最早由Ross等提出,提取液采用凝胶渗透层析(GPC)净化(SX-3柱),杀菌剂的回收率超过90%。1989年,瑞典国家食品管理局将其列为国家多残留分析方法,替代了原来由Anderson和Ohlin建议的方法。现在该方法已能检测约160种农药、异构体及降解代谢产物。在荷兰的国家方法中,乙酸乙酯也作为主要的提取溶剂。由于省去净化步骤,乙酸乙酯提取方法也被称为在线提取法(on-line extraction methods),其理论基础是Gibbs三角,可用于在线提取的有机溶剂对还有正己烷/丙酮(8∶2)、乙酸乙酯/二甲苯、丙酮或乙腈/二氯甲烷或石油醚等。2.1.4其它提取方法固相萃取是近年发展较快的一种提取、净化方法,作为一种提取技术,主要应用于液体样品中农药的提取,用于农产品及土壤等固体或半固体样品中农药的提取,实质上是一种净化、富集过程。商品化的固相萃取装置很多,主要是固相萃取小柱、固相萃取盘等,其发展主要体现在填料方面,如石墨化炭黑、键合硅胶、弗罗里硅土、活性炭、硅胶等。基质固相分散萃取是类似于固相萃取的一种提取、净化、富集技术,其是将吸附剂或填料与样品一起研磨分散,然后装柱,用有机溶剂淋洗,使农药淋洗出来,淋洗液一般无需进一步净化,可直接进[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]或高效液相色谱检测。MSPD实际上将包括样品匀浆、细胞裂解、完全提取、分馏及纯化过程集于一个简单的过程。有些淋出杂质较多的样品,也可进行净化、分析。Kadenski等采用MSPD技术,研究了多种农药在一些蔬菜、水果中的回收。与传统方法相比,MSPD具有显著的优点,主要表现在,缩短了样品分析周期;减少了样品量,从而极大的降低了溶剂的用量,降低了环境污染的可能性并提高了操作安全性;适用于自动化操作等。
顶空气相色谱法(HS-GC)已经被制药企业的实验室采用了很多年,但是人们尚未找到过一种挥发性有机物杂质背景值含量极低的溶剂。最近几年,随着检测器的灵敏度不断的增加,残留溶剂最小量的控制要求也越来越严格,所以寻找一种高质量并且适用于HS-GC-FID/HS-GC-MS分析的溶剂成为大势所趋。 在药物生产过程中,无论是原材料还是成品,残留溶剂的分析都是十分必要的。与样品其他组分相比,残留溶剂沸点具有沸点低、易挥发等特点,最合适的分析方法便是使用氢火焰离子化检测器(FID)或者质谱检测器(MS)的顶空气相色谱法。样品需要被溶解于一种无残留溶剂的高沸点溶剂。样品在高温下被孵化,残留溶剂就能以气态和样品的其他组分分离,从而进行HS-GC-FID/HS-GCMS。 气相色谱顶空溶剂中如甲醇、乙腈、乙醇、异丙醇、正丙醇、正丁醇、环己烷、正己烷、正庚烷、二恶烷、二氯甲烷、吡啶、四氢呋喃、叔丁基甲醚、乙酸乙酯、乙酸丁酯、乙酸异丙酯、苯系物(甲苯、乙苯、二甲苯)等数十种有机挥发性化合物杂质背景值极低,均低于1ppm。产品货号 产品名称 品牌 规格4.109001.1000 气相顶空级N,N-二甲基甲酰胺,DMF,for HS-GC CNW 1L 4.109002.1000 气相顶空级N,N-二甲基乙酰胺,DMA,for HS-GC CNW 1L 4.109003.1000 气相顶空级二甲基亚砜,DMSO,for HS-GC CNW 1L 4.109004.1000 气相顶空级1,3-二甲基-2-咪唑啉酮,DMI,for HS-GC CNW 1L 4.109005.1000 气相顶空级N-甲基吡咯烷酮,NMP,for HS-GC CNW 1L 4.109006.1000 气相顶空级环己酮,for HS-GC CNW 1L 4.109008.1000 吹扫捕集级甲醇,for purge and trap CNW 1L 4.109007.1000 气相顶空级水,for HS-GC CNW 1L
一、液相色谱法GB 29694-2013 食品安全国家标准 动物性食品中13种磺胺类药物多残留的测定 高效液相色谱法GB/T 18932.5-2002 蜂蜜中磺胺醋酰、磺胺吡啶、磺胺甲基嘧啶、磺胺甲氧哒嗪、磺胺对甲氧嘧啶、磺胺氯哒嗪、磺胺甲基异恶唑、磺胺二甲氧嘧啶残留量的测定方法 液相色谱法GB/T 19542-2007 饲料中磺胺类药物的测定 高效液相色谱法GB/T 8381.10-2005 饲料中磺胺喹噁啉的测定 高效液相色谱法SB/T 10388-2004 畜禽肉中磺胺二甲嘧啶、磺胺甲恶唑的测定SN/T 1965-2007 鳗鱼及其制品中磺胺类药物残留量测定方法 高效液相色谱法农业部1025号公告-15-2008 鸡蛋中磺胺喹噁啉残留检测高效液相色谱法农业部1486号公告-7-2010 饲料中9种磺胺类药物的测定 高效液相色谱法农业部1879号公告-2-2012 饲料中磺胺氯吡嗪钠的测定 高效液相色谱法农业部958号公告-12-2007 水产品中磺胺类药物残留量的测定 液相色谱法DB33/T 701-2008 配合饲料中磺胺类药物的测定 高效液相色谱法DB34/T 1033-2009 动物组织中地克珠利和磺胺类药物的残留测定-高效液相色谱法DB34/T 1034-2009 动物组织中呋喃唑酮和磺胺类药物的残留测定-高效液相色谱法二、液相色谱-串联质谱法GB/T 18932.17-2003 蜂蜜中16种磺胺残留量的测定方法 液相色谱-串联质谱法GB/T 20759-2006 畜禽肉中十六种磺胺类药物残留量的测定 液相色谱-串联质谱法GB/T 21316-2007 动物源性食品中磺胺类药物残留量的测定 高效液相色谱-质谱/质谱法GB/T 22947-2008 蜂王浆中十八种磺胺类药物残留量的测定 液相色谱-串联质谱法GB/T 22951-2008 河豚鱼、鳗鱼中十八种磺胺类药物残留量的测定 液相色谱-串联质谱法GB/T 22966-2008 牛奶和奶粉中16种磺胺类药物残留量的测定 液相色谱-串联质谱法SN/T 2580-2010 进出口蜂王浆中16种磺胺类药物残留量的测定 液相色谱-质谱/质谱法农业部1025号公告-23-2008 动物源食品中磺胺类药物残留检测 液相色谱-串联质谱法农业部1077号公告-1-2008 水产品中17种磺胺类及15种喹诺酮类药物残留量的测定 液相色谱-串联质谱法农业部781号公告-12-2006 牛奶中磺胺类药物残留量的测定 液相色谱-串联质谱法DB13/T 1384.10-2011 饲料中20种磺胺类药物的测定DB33/T 746-2009 动物源性食品中20种磺胺类药物残留量的测定 液相色谱-串联质谱测定法三、酶联免疫吸附法DB51/T 470-2005 鸡蛋中磺胺二甲嘧啶残留检测方法-酶联免疫吸附测定(ELISA)法SN/T 1960-2007 进出口动物源性食品中磺胺类药物残留量的检测方法 酶联免疫吸附法农业部1025号公告-24-2008 动物源食品中磺胺二甲嘧啶残留检测 酶联免疫吸附法农业部1025号公告-7-2008 动物性食品中磺胺类药物残留检测 酶联免疫吸附法四、放射受体分析法GB/T 21173-2007 动物源性食品中磺胺类药物残留分析法 放射受体分析法SN/T 1765-2006 动物组织中磺胺类抗生素残留量检测方法 放射免疫受体筛选法SN/T 2799-2011 进出口蜂王浆中磺胺类药物残留量测定方法 放射受体分析法五、气相色谱法SN 0498-1995 出口肉类中磺胺间二甲氧嘧啶残留量检验方法







 我要推广仪器
我要推广仪器
 下载APP
下载APP