甲基化成肿瘤检测新靶标?五种新型DNA甲基化酶检测技术进展揭秘
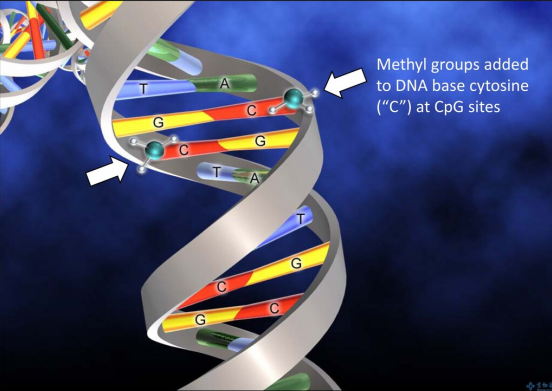
DNA甲基化是哺乳动物基因组中最常见的表观遗传事件之一,即DNA中核苷酸与甲基基团的共价修饰[2]。DNA甲基化与人的生命进程有着密不可分的关系。细胞的增殖与分化、染色体完整性的维护或者X染色体的活性等等都离不开DNA甲基化的控制,DNA甲基化流程在胚胎发育中是无处不在的[1]。如果DNA甲基化进程出现异常,会导致生物体出现各种各样的疾病以及身体的生长缺陷或生理紊乱。DNA与蛋白质之间的相互作用如果出现异常,会影响基因的表达,从而引起人体内肿瘤的发生或者肿瘤的转移,这一切的源头都是DNA甲基化进程出现异常的结果[3]。DNA甲基化酶是肿瘤治疗靶点DNA甲基化酶是一种修饰酶,经常与限制性内切酶一同出现。在真核生物基因组以及原核生物基因组中,普遍存在DNA甲基化酶维持以及催化DNA甲基化过程的现象。DNA甲基化酶被广泛认为是一种治疗靶点以及预测生物甲基化过程的标志物,在单细胞水平上准确灵敏地检测DNA甲基化酶对于肿瘤医学上的临床诊断以及临床治疗甚至是生物学研究有着至关重要的作用。根据甲基化的核苷酸和位置被分为三组,即腺嘌呤的甲基化、胞嘧啶的4-N甲基化和胞嘧啶的5-C甲基化。所有已知的DNA甲基化酶在其甲基化过程中以s-腺苷甲硫氨酸作为甲基供体。最常见的DNA甲基化不仅发生在胞嘧啶嘧啶环5-C位置的CpG位点上,还发生在对称四核苷酸5’-G-A-T-C-3’ 中腺嘌呤环的6-N位置[4,5]。传统DNA甲基化酶检测方法有局限 DNA甲基化酶活性的高灵敏度检测在基因调控、表观遗传修饰、临床诊断和治疗等方面具有重要意义。传统用于检测DNA甲基化酶活性的方法包括高效液相色谱法(HPLC)[6], 聚合酶链反应(PCR)[7],凝胶电泳[8],高效毛细管电泳(HPCE)[9],以及使用同位素标记的s-腺苷甲硫氨酸甲基化检测[10,11]。尽管这些技术在实验室实践中被证明是有用的,但它们具有局限性。例如,大多数技术不仅使用笨重昂贵的设备,而且需要复杂的样品制备和数据分析所需的大量时间。同位素标记等技术是有效的,但它们往往需要费力的样品制备、同位素标记、复杂的设备和大量的DNA,使得它们不适合在医护点使用。所以,DNA甲基化酶活性检测迫切需要简单、便携、高灵敏度和低成本的检测方法。在最近的技术进步中,许多替代的DNA甲基化酶活性测定方法,如放射法、比色法、荧光法、电化学法等已被提出。此外,其中许多与纳米材料或酶结合,以显著提高它们的敏感性。放射法、蛋白质纳米孔等新型检测技术兴起 放射法:同位素标记作为最早检测DNA甲基化酶活性的方法之一,早期广泛应用于检测DNA甲基化酶和DNA甲基化的活性[12,13]。在由DNA甲基化酶催化的甲基化过程中,同位素标记的甲基部分转移到DNA上,从而赋予甲基化的DNA放射性。这种放射性可以很方便地用闪烁计数器或放射自显像仪来检测。可惜的是,放射性试剂的介入是限制这种试验在中央实验室进行的最大缺点。对无辐射DNA甲基化酶活性检测的研究导致了甲基化特异性PCR[14]、HPCE[9]和HPLC等替代品的发展[7,14],而甲基化特异性PCR被认为是较好的方法。尽管非放射性,上述DNA甲基化酶活性检测需要庞大且通常昂贵的设备,冗长且耗时的样品制备和数据分析,以及繁琐的检测方案,这在临床实践中也比较难以实现全覆盖。比色法:比色法用于DNA甲基化酶活性检测依赖于颜色变化的目视观察或与DNA甲基化酶相关的吸收光谱的光谱测量。它们具有成本低、简单、可移植性和在某些情况下无需仪器的优点。虽然紫外-可见光谱法可以量化DNA,但甲基化和未甲基化DNA在紫外-可见吸收特性上的低灵敏度和不显著差异基本否定了紫外-可见光谱法直接检测DNA甲基化酶活性[15~17]。金纳米粒子:金纳米粒子(AuNPs)由于其表面的等离子体共振吸收的高消光系数且强依赖于粒子间距离,在DNA甲基化酶活性检测的比色法研究中引起了广泛关注。如图1 所示,金纳米粒子表面包覆有双链DNA (ds-DNA),其中一条链包含DNA甲基化酶识别序列和5’-硫醇末端。在DNA甲基化酶存在的情况下,如图1 B 所示,DNA甲基化酶被共价标记在ds-DNA中碱基环的6-C位置,因为在5-N位置缺乏一个质子阻止了β-消除,甲基化的DNA不能被核酸外切酶 ExoⅠ剪切,因此金纳米粒子仍然均匀地分散在溶液中 [18]。从而实现DNA甲基化酶活性的检测。结果表明,在526 nm处,金纳米粒子聚集物的吸光度与DNA甲基化酶的活性呈2 ~ 32 U / mL的线性关系,检出限为0.5 U/ mL。图1. (A)基于ABP的比色生物传感器的示意图(B) DNA甲基化酶的检测机制 荧光法:荧光指吸收激发荧光团的光,以促进电子从基态到激发态,电子迅速地回到激发态的最低能级,然后当电子最终返回基态时,发出波长较长的光。与其他DNA甲基化酶活性测定法相比,荧光法检测DNA甲基化酶活性的优点是检测过程简单,灵敏度高,但其复杂的光学性能限制了其在集中实验室的应用[19~20]。图2. 基于外切酶的靶循环的DNA甲基化酶活性检测原理图电化学法:电化学生物分析技术的发展一直是现代分析化学研究的热点之一。电化学法用于DNA甲基化酶分析包括测量电流、电压、电荷和电阻等电量,以反映DNA甲基化酶的活性。与许多其他类型的DNA甲基化酶活性的检测相比,它们具有低成本、高灵敏度、执行现场监测的能力以及非常适合微型化和集成微制造技术的优点[22~23]。Zhi-Qiang Gao等人在2014年报道了一种简单、高灵敏度的DNA甲基化酶电化学活性测定方法。该方法采用电催化氧化抗坏血酸(AA)的信号放大手段,通过一个螺纹插层N,N -2(3-丙基咪唑)-1,4,5,8-萘二酰亚胺(PIND)电催化氧化还原Os(bpy)2Cl+ (PIND-Os),包含5’-CCGG-3’ 对称序列的ds-DNA首先固定在金电极上。然后用DNA甲基化酶孵育电极,经过酶催化特定CpG二核苷酸的甲基化,然后用识别5’-CCGG-3’ 序列的限制性内切酶 Hpa II 剪切酶处理电极,从而实现DNA甲基化酶活性检测的目的[24]。图3. DNA甲基化酶活性的检测原理示意图蛋白质纳米孔:蛋白质纳米孔检测技术是在单分子水平上以低成本、无标签和高通量的方式研究生物分子的检测技术。近年来,纳米孔技术正从生物传感的角度进行研究[25]。应用于核酸特征鉴定、化学反应过程的测量、蛋白质分析、疾病相关蛋白状态的检测以及酶动力学的研究等[26]。α-溶血7素是一种蛋白质纳米孔,它自发地插入到脂质双层膜中,形成一个纳米孔[27]。当一个带电分子在外加电势下通过蛋白质纳米孔时,它会引起离子电流的瞬态变化,电流变化事件被记录下来。被分析物可以通过当前电流发生的频率进行量化,特征电流信号则可以揭示被分析物的各种特征[28~30]。该检测方法不需要对DNA探针进行任何化学修饰,既方便又节约成本,减少了样品消耗。 图4. 用于分析DNA甲基化酶活性的纳米孔试验的示意图 在过去的十几年中,DNA甲基化酶活性的检测取得了重大进展。有几种方法有希望可在临床检测,使得该方法在用于癌症诊断、预后和治疗方面显示出了希望。比色法依赖于颜色变化的目视观察或与DNA甲基化酶相关的吸收光谱的光谱测量,具有成本低、简单、可移植性和在某些情况下无需仪器的优点,但是检出限相对较高。荧光法检测DNA甲基化酶活性的检测过程简单,检出限相对理想,但其复杂的光学性能以及昂贵的仪器设备限制了其在生活中的应用。电化学法由于需要构建较复杂的反应电极材料而使得其在临床上受到了一定的限制。蛋白质纳米孔的检测方法不需要对DNA探针进行任何化学修饰,既方便又节约成本,减少了样品消耗,检出限相对较为理想,并且已经成功应用于人类血清样本。这类检测可能最终为常规DNA甲基化酶活性的检测和分子诊断打开大门,为疾病的管理和诊断带来新的前景。 作者:王家海、骆 乐 作者简介:王家海,博士,教授,硕士生导师/博士生导师,广州大学化学化工学院;分析化学专业;主要研究领域为“基于核算纳米结构为信号传导载体的纳米孔传感器”;在核酸探针和仿生纳米孔两方面开展了一系列分子识别的工作,也为将来进一步开展分析化学研究打下了坚实的基础,期间积累了多种前沿分析方法和技术:仿生纳米孔制备和检测;微纳米加工技术;核酸探针人工合成技术。参 考 文 献 [1] 陈晓娟,闫少春,邵国,等.人DNA甲基化转移酶的分类及其功能[J].包头医学院学报,2014,30(04):136-138.[2] Das PM, et al. DNA methylation and cancer[J]. Clin. Oncol. 2004 22: 4632-4642.[3] Jurkowska RZ, et al. Structure and function of mammalian DNA methyltransferases[J]. ChemBioChem 2011 12: 206-222.[4] Lee GE, et al. DNA methyltransferase 1-associated protein (dmap1) is a co-repressor that stimulates DNA methylation globally and locally at sites of double strand break repair[J]. Biol. Chem. 2010 285: 37630-37640.[5] Liu SN, et al. Assay Methods of DNA Methylation and Their Applications in Cancer Diagnosis and Therapy[J]. Chinese J.Anal. Chem. 2011 39: 1451-1458.[6] Boye E, et al. Quantification of dam methyltransferase in Escherichia coli[J]. Bacteriol. 1992 174: 1682-1685.[7] Eads CA, et al. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression[J]. Cancer Res. 1999 59: 2302-2306.[8] Bergerat A, et al. Allosteric and catalytic binding of s-adenosylmethionine to escherichia coli DNA adenine methyltransferase monitored by 3H NMR[J]. Proc. Natl. Acad. Sci. U. S. A. 1991 88: 6394-6397.[9] Fraga MF, et al. Rapid quantification of DNA methylation by high performance capillary electrophoresis[J]. Electrophoresis 2000 21: 2990-2994.[10] Yokochi T, et al. DMB (dnmt-magnetic beads) assay: measuring DNA methyltransferase activity in vitro[J]. Methods Mol. Biol. 2004 287: 285-296.[11] Adams RLP, et al. Microassay for DNA methyltransferase[J]. Biochem. Bioph. Methods 1991 22: 19-22.[12] Jurkowska RZ, et al. DNA methyltransferase assays[J]. Methods Mol. Biol. 2011 791: 157-177.[13] Pradhan S, et al. Recombinant human DNA (cytosine-5) methyltransferase [J]. Biol. Chem. 1999 274: 33002-33010.[14] Herman JG, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands[J]. Proc. Natl. Acad. Sci. U. S. A. 1996 93: 9821-9826.[15] Kattenhorn, L. M. Korbel, G. A. Kessler, B. M. Spooner, E. Ploegh, H. L. Mol. Cell 2005, 19, 547−557.[16] Mosammaparast, N. Shi, Y. Annu. Rev. Biochem. 2010, 79, 155−179.[17] Barglow, K. T. Cravatt, B. F. Angew. Chem., Int. Ed. 2006, 45, 7408−7411.[18] Wu Z, et al. Activity-based DNA-gold nanoparticle probe as colorimetric biosensor for DNA methyltransferase/glycosylase assay[J]. Anal. Chem. 2013 85: 4376-4383.[19] Zhu, C. Wen, Y. Peng, H. Long, Y. He, Y. Huang, Q. Li, D. Fan, C. Anal. Bioanal. Chem. 2011, 399, 3459−3464.[20] Chen, F. Zhao, Y. Analyst 2013, 138, 284−289.[21] Xing XW, et al. Sensitive detection of DNA methyltransferase activity based on exonuclease-mediated target recycling[J]. Anal. Chem. 2014 86: 11269-11274.[22] Wu, H. Liu, S. Jiang, J. Shen, G. Yu, R. Chem. Commun. 2012, 48, 6280−6282[23] Wang, M. Xu, Z. Chen, L. Yin, H. Ai, S. Anal. Chem. 2012, 84, 9072−9078[24] Deng H, et al. Highly sensitive electrochemical methyltransferase activity assay[J]. Anal. Chem. 2014 86: 2117-2123.[25] Howorka, S. Siwy, Z. Nanopore Analytics: Sensing of Single Molecules. Chem. Soc. Rev. 2009, 38, 2360−2384.[26] Song, L. Hobaugh, M. R. Shustak, C. Cheley, S. Bayley, H. Gouaux, J. E. Structure of Staphylococcal α-Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859−1865.[27] Lin, L. Yan, J. Li, J. Small-Molecule Triggered Cascade Enzymatic Catalysis in Hour-Glass Shaped Nanochannel Reactor for Glucose Monitoring. Anal. Chem. 2014, 86, 10546−10551.[28] Li, J. Yan, H. Wang, K. Tan, W. Zhou, X. Anal. Chem. 2007, 79, 1050−1056.[29] Wood, R. J. Maynard-Smith, M. D. Robinson, V. L. Oyston, P. C. F. Titball, R. W. Roach, P. L. PLoS One 2007, 2, e801−e801.[30] Wood, R. J. McKelvie, J. C. Maynard-Smith, M. D. Roach, P. L. Nucleic Acids Res. 2010, 38, e107−e107.[31] Jinghong Li, et al. Nanopore-based, label-free, and real-time monitoring assay for DNA methyltransferase activity and inhibition[J]. Anal. Chem. 2017 89: 13252−13260.

























 我要推广仪器
我要推广仪器
 下载APP
下载APP