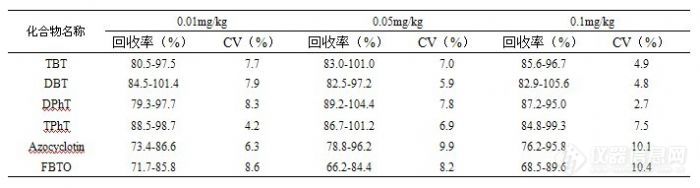
在线凝胶渗透色谱-气相色谱-质谱法测定茶叶中多种有机锡化合物残留有机锡化合物(OTCs)是一类包含C-Sn共价健的化合物,主要用于聚氯乙烯聚合物的稳定剂、化学反应中的催化剂及农业生产中的除草剂、灭菌剂和杀虫剂。由于有机锡化合物在工农业生产中的广泛应用,造成其在环境中的普遍污染,并沿着食物链的不断传递而对人体健康造成影响。作为一种重要的内分泌干扰物质,浓度在ng/L水平的有机锡化合物即可对水环境中的生物产生毒性危害。因此,有机锡化合物的痕量与超痕量分析是当今环境和食品安全分析领域的重要研究课题。本文采用在线凝胶色谱串联气相色谱-质谱(GPC-GC-MS)技术,结合大体积进样,同时检测茶叶中的6种有机锡化合物,具有检出限低、快速、简便、重复性好等优点及较好的实际应用价值。1. 实验部分1.1 仪器与试剂 GPC-GC/MS QP 2010 Plus在线凝胶渗透色谱-气相色谱质谱联用仪(日本岛津公司),配有程序升温(PTV)进样口和EI电离源;Envi-Carb活性碳固相萃取小柱(250 mg,3 mL,Supelco公司);Florisil固相萃取小柱(250 mg,3 mL,Supelco公司);快速混匀器(SK-1型,常州澳华仪器有限公司);氮吹仪(KL 5121型);低速离心机(10000 rpm以下,TDZ 4-WS型);电子分析天平(PB203-N型)。丙酮、环己烷和正己烷均为HPLC级试剂;无水乙醚、H2SO4为分析纯;无水Na2SO4为分析纯,650℃灼烧4 h,贮于密封干燥器中备用。所用水均为超纯水。六种标准品:二丁基二氯化锡([font=Ti
有机氟化合物organic fluorine compound有机化合物分子中与碳原子连接的氢被氟取代的一类元素有机化合物。分子中全部碳-氢键都转化为碳-氟键的化合物称全氟有机化合物,部分取代的称单氟或多氟有机化合物。由于氟是电负性最大的元素,多氟有机化合物具有化学稳定性、表面活性和优良的耐温性能等特点。有机氟化合物分为以下几类:①含氟烷烃。氟利昂是氟化的甲烷和乙烷,也可以含氯或溴。这类化合物多数为气体或低沸点液体,不燃,化学稳定,耐热,低毒。主要用作制冷剂、喷雾剂等,最常用的是氟利昂-11(CFCl3)和氟利昂-12(CF2Cl2)。这类化合物也是重要的含氟化工原料或溶剂。如二氟氯甲烷用于合成四氟乙烯;1,1,2-三氟三氯乙烷用于合成三氟氯乙烯,也是优良的溶剂。含氟碘代烷如三氟碘甲烷等为重要的合成中间体。一些低分子含氟烷烃和含氟醚具有麻醉作用,并有不燃、低毒的优点,可用作吸入麻醉剂,例如1,1,1-三氟-2-氯-2-溴乙烷(俗称氟烷)已广泛用于临床。②含氟烯烃。以四氟乙烯、偏氟乙烯和三氟氯乙烯等为代表。四氟乙烯为最主要的含氟单体,可以聚合成聚四氟乙烯,或与其他单体共聚合成多种含氟高分子。偏氟乙烯CF2=CH2在空气中的浓度在5.8%~20.3%之间时,遇火可爆炸,主要用于与其他单体共聚合制取含氟弹性体。三氟氯乙烯主要作为单体,用于合成均聚物或共聚物。③含氟芳烃。苯分子中的氢可以通过间接方法部分或全部用氟取代。氟苯为含氟芳烃的代表。多氟苯或全氟苯易与亲核试剂发生取代反应。
2011年10月,上海安谱公司协同美国REGIS科技公司拜访了上海诸多知名手性应用实验室,如睿智化学和药明康德等手性分析领域的领军企业,并和用户进行了深入交流。美国REGIS 科技公司成立于1956年,在化合物的手性分离上一直处于世界的领先地位,其公司的主要手性柱类型包括: 美国REGIS 科技公司成立于1956年,在化合物的手性分离上一直处于世界的领先地位,其公司的主要手性柱类型包括: 1. Pirkle类型固定相(Whelk-O 1,Whelk-O 2,Leucine,DACH-DNB,ULMO等) 2. 多糖类固定相(RegisPack,RegisCell,RegisPack CLA-1) 3. 冠醚类固定相(ChiroSil (RCA)(+) 和 (SCA)(-),专用于氨基酸分析) 其中只需Whelk-O,RegisPack,RegisCell三款类型就可以分离95%的手性化合物。 Whelk-O固定相是REGIS非常独特的,具有不可替代性,其优点包括: ● 适用于多种化合物组分的手性分离,应用范围广泛; ● 柱子耐用性强,可耐受多种强溶剂; ● 具有转变洗脱顺序的能力; ● 出色的色谱分离效率; ● 具备从分析级到制备级各种规格手性柱。 是不是就是说手性的分离以后是特别的容易呢??和极限的相比又如何?都介绍自己的产品好啊,以后要不购买上做个对比???http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif安谱专家能否做个更详细的介绍呢??
气相色谱进行多种化合物混合溶液分析时,时常会出现大多数化合物的保留时间及响应重现性都很好,其中某一种或几种化合物重现性较差,遇到这种情况该如何解决呢?
如题,例如MRM扫描做3个化合物,有2个化合物的峰重叠了,尝试了多种方法分离(该流动相比例啊,梯度洗脱啊,换长柱啊),但都达不到完全的理想分离效果,这样会影响定量吗?
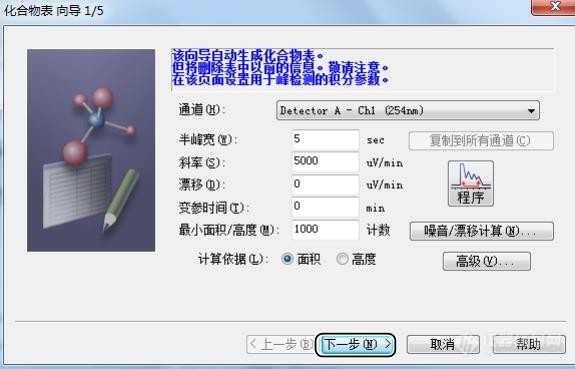
[align=left][font=宋体]样品进样完成后,最终都要处理数据。这时候我们需要筛选出对应的色谱峰,制作对应的曲线,得出正确的结果。[/font][/align][font=宋体]岛津软件需要制作化合物表,我们筛选出正确的色谱峰添加到化合物表中,方法有种方法。[/font][font=宋体]第一种可以使用化合物向导,中间可以进行积分、峰识别以及将定量处理参数输入到连[/font][font=宋体]续窗口中。[/font][font=Calibri] [img=,575,369]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030933375259_1609_5979722_3.jpg!w575x369.jpg[/img][/font][font=宋体][font=宋体]点击左侧的向导,半峰宽、斜率、漂移、最小面积等这些参数(这些参数的设置都是为了排除不需要的色谱峰,比如最小面积设置为[/font][font=Calibri]1000[/font][font=宋体],则面积小于[/font][font=Calibri]1000[/font][font=宋体]的色谱峰就不进行积分)基本都可以默认,点击下一步。[/font][/font][font=Calibri] [img=,576,366]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030933458986_2400_5979722_3.jpg!w576x366.jpg[/img][/font][font=宋体]这时候参考标准物质的出峰时间,勾选对应保留时间的色谱峰。继续下一步[/font][font=宋体] [img=,574,356]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030933543930_8576_5979722_3.jpg!w574x356.jpg[/img][/font][font=宋体]选择正确的定量方法、浓度、级别数等。继续下一步[/font][font=宋体] [img=,576,369]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934028286_5423_5979722_3.jpg!w576x369.jpg[/img][/font][font=宋体]然后设置识别方法、窗宽以及其他参数等。继续下一步[/font][font=宋体] [img=,569,359]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934109321_2525_5979722_3.jpg!w569x359.jpg[/img][/font][font=宋体]进入化合物表,设置对应的浓度等信息,点击完成。[/font][font=宋体] [img=,569,359]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934197922_3795_5979722_3.jpg!w569x359.jpg[/img][/font][font=宋体]最终应用到方法。[/font][font=宋体] [/font][font=宋体]第二个方法,直接打开单个数据文件,用鼠标右键单击所需峰的峰号,然后单击将选择峰记录到化合物表。[/font][font=Calibri][font=宋体]在方法视图里单击化合物标签。在化合物表中输入登录峰的化合物名和浓度[/font][/font][font=宋体],在显示峰定位线的时候,使用鼠标可简单设置化合物表内的保留时间。最终应用到方法。[/font][font=Calibri] [img=,690,465]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934352396_4572_5979722_3.jpg!w690x465.jpg[/img][img=,563,457]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934463920_6334_5979722_3.jpg!w563x457.jpg[/img][/font][font=宋体]第三个方法,直接[/font][font=Calibri][font=宋体]在方法[/font][/font][font=宋体]编辑[/font][font=Calibri][font=宋体]里单击化合物标签。在化合物表中[/font][/font][font=宋体]手工[/font][font=Calibri][font=宋体]输入登录峰的化合物名和浓度[/font][/font][font=宋体],在显示峰定位线的时候,使用鼠标可简单设置化合物表内的保留时间。最终应用到方法。[/font][font=宋体] [img=,563,457]https://ng1.17img.cn/bbsfiles/images/2023/10/202310030934463920_6334_5979722_3.jpg!w563x457.jpg[/img][/font][font=宋体]这三种方法各有特点,大家可以灵活运用。[/font][font=宋体] [/font][font=Calibri] [/font][font=Calibri] [/font]
[font=SimSun, STSong, &]检测结果是碳水化合物为0,请问我标签印刷刻意强调无糖还需要在碳水化合物下面一栏标注糖为0吗?[/font][font=SimSun, STSong, &]1、如果不需要标注,那打假人员说依据GB7718 -2011 4.1.4.2 如果在食品的标签上特别强调一种或多种[/font][font=SimSun, STSong, &]配料或成分的含量较低或无时,应标示所强调配料或成分在成品中的含量。你宣称无糖,未在配料或[/font][font=SimSun, STSong, &]成品中标示含量。[/font][font=SimSun, STSong, &]2、如果在碳水化合物下面标注糖为0,那我们没有检测糖为0啊,但是实际你碳水化合物为0,那糖肯[/font][font=SimSun, STSong, &]定为0[/font]
各位大虾好,我们要检测盒子(PE、PP、聚碳酯质材等)上的微量硅油、DOP(邻苯二甲酸二辛酯)、氨基化合物等,用正己烷洗盒子,蒸至剩少许用毛细管在KBr上点样,进行FT-IR定性检测。但由于硅油、DOP、氨基化合物很微量,做出的谱图很杂,硅油、DOP、氨基化合物的特征峰很难分辨,还有很多其他未知峰,很难判断盒子上是否含硅油、DOP、氨基化合物。请教各位大虾,用红外对一种样品里含有的多种微量物质来定性,且本底物质杂,这种方法是否可行?是不是有必要先进行;液相或[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分离后再运用合适仪器来定性?谢谢各位。
近日,中国农业科学院烟草研究所植物功能成分与综合利用创新团队在烟草内生真菌中发现了抑菌、杀虫活性显著且毒性较小的异戊烯基化吲哚类活性化合物,为具有自主知识产权的高效低毒生物农药的研发提供了模板化合物。相关研究成果在线发表在《农业与食品化学杂志(Journal of Agricultural and Food Chemistry)》。 据张鹏副研究员介绍,传统化学合成农药在为农业生产带来巨大经济效益的同时,也对生态系统造成了一系列弊端。微生物源农药因具有高效低毒、环境友好等特点,在植物病虫害防治中的作用日益明显。植物功能成分与综合利用创新团队从一株烟草来源内生真菌接骨木镰刀菌TE-6L中分离获得6个异戊烯基化吲哚类代谢产物,其中包括2个新结构化合物。研究表明,该类代谢产物能够显著抑制多种植物病原菌并具有杀虫活性;同时,该团队以斑马鱼胚胎为模型,首次评估了该类化合物的发育毒性。该类化合物结构新颖、活性显著且毒性较低,具有开发成为新的生物农药的潜力。 该研究得到国家自然科学基金和中国农科院科技创新工程资助。
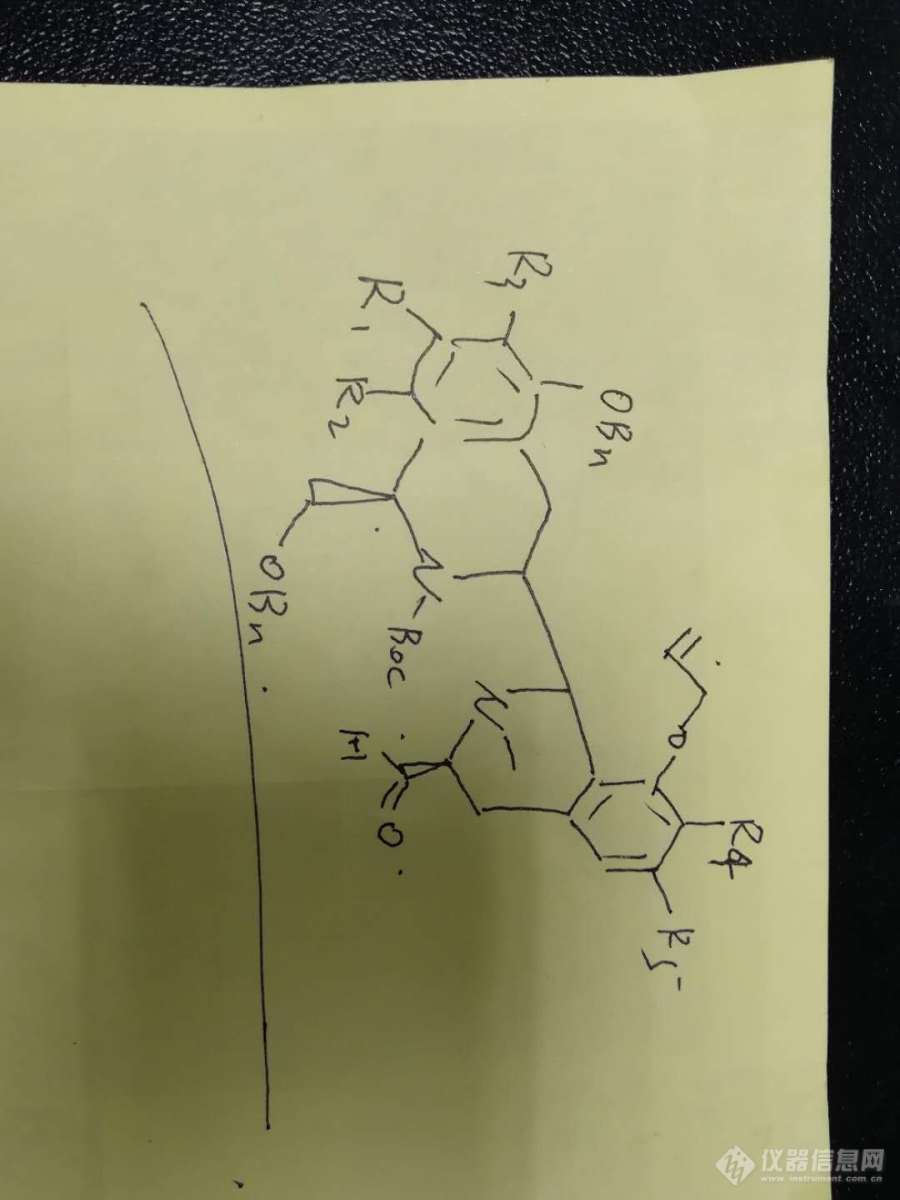
一个未知 的复杂化合物,查不到文献,以下为其大概的结构式,分子量约700多,以下尝试了几种方法、[img=,401,312]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131304270646_2129_3116636_3.png!w401x312.jpg[/img]刚开始用的溶剂是乙腈,体系为TFA乙腈体系,无法得到理想的峰形,而且尝试了很多种都不可以,后来尝试可一个梯度,这个梯度下出峰尖锐,但是并不理想,有峰未分离开来。(这个梯度是尝试了很多种后选择的分离度相对较好的条件。)梯度:0 50% 50% 5 50% 50% 9 15 % 85% 17 15% 85% 19 5% 95% 27 5% 95% 27.01 50% 50%后来尝试往有机相加了一定比例的甲醇,约30%(尝试过15-45%)基本差不多。乙腈溶解,0.1%TFA和30%+70%乙腈做流动相,梯度跟上图一样,出了两个峰,但实验员反映,打过核磁没有发现同分异构体也没有其余杂质。如下图[img=,690,345]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131309278474_7582_3116636_3.png!w690x345.jpg[/img]后来怀疑会跟流动相中的甲醇反应,于是将溶剂换成了甲醇,梯度流动相与上图一致,发现峰变成了一个,全部转化为左边峰,证明产品会与甲醇反应。(也试过异丙醇,一样会反应)如下图。[img=,690,353]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131312161097_8267_3116636_3.png!w690x353.jpg[/img]因此这个体系不可取了,实验员认为TFA可能也会与化合物反应,并且猜测化合物为中性,让用纯水,但是在我看来中性化合物的话,PH的影响对其在液相中的保留时间分离度不会有明显变化,但是还是尝试了一下纯水体系。乙腈溶解,纯水和乙腈作为流动相,梯度与上面相同,保留时间延后,分离度变好。但是左边依然有一个峰,这与最开始TFA和乙腈体系下有点相似,左边有一个未分离开来的小峰,在这个体系下分开了。那么说明这个化合物并非是中性化合物吗?如下图[img=,690,351]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131317512074_2363_3116636_3.png!w690x351.jpg[/img]为了证明左边的峰是否是与甲醇反应的峰一致,于是又进了一针甲醇溶解的,梯度进样量与上相同。结果变成了三个峰,第三个峰变小了,中间的峰变高,并且多了第一个峰……[img=,690,339]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131325256497_9234_3116636_3.png!w690x339.jpg[/img]就以上几种尝试多了很多无法想通的问题:1、这个化合物到底是偏中性还是碱性。2、在TFA体系下,甲醇溶解后进样只出一个峰,而在纯水体系下,却出了三个峰,那么是否说明起始在TFA的体系下也有三个峰,只是未能完全分开。3、谁做过类似的化合物,是否有较好的分析方法呢?最新的情况:还有一个化合物跟上面的相似,是其前一个步骤称为化合物1,只有一个基团不同,就是右下角是羟基,其余一样,上面那个就称为化合物2然后我们用上面的方法,TFA体系和纯水体系,有机相都为乙腈的条件下走了两针。在TFA体系下峰形良好,出峰时间约18.7,在中性体系下,峰形宽胖,出峰时间15.0min。然后呢还得知了其PKA值为6.7左右,上面那个化合物2PKA值5.3左右。那么为了更好的分离这两个物质,我选择的流动相应该PH在多少?已知TFA/磷酸这种较弱的PH下不可以分离,两个化合物峰靠的很近。在纯水体系下,两个化合物分离的很远,带羟基的化合物1出峰时间15.0min,但是峰形宽胖,带醛基的化合物2出峰时间27min,分离度良好,但是时间太靠后,试了好多其他梯度都没办法解决。我准备尝试0.1%的氨水体系,不知道是否可以。有没有其他更好的调节方式呢?望指教~~~
安捷伦chemsation的RTL功能一个方法只能锁定一种化合物吗?若能同时锁定多种化合物,是不是对于每种化合物都要在5种不同压力下验证?这样的话岂不是非常繁琐?
我想问个比较常用的问题就是酸性化合物和碱性化合物怎样判断。因为我根据PKA好像是判断不出化合物的酸碱性。我有三个疑问如下1.三聚氰胺是一个碱性化合物他的pka=8,苯酚是一个酸性化合物但是他的pka是9.99.按照pka来划分有机化合物的酸碱性不是很合适。有没有高手帮忙解惑一下怎么判断有机化合物的酸碱性?以克球粉为例因为克球粉带N,所以我感觉克球粉是一个碱性化合物但是他是一个酸性的。结构式如下:http://www.ichemistry.cn/pstructure/2971-90-6.png2.还有一句话是在液相色谱柱应用的时候经常说的,在C18上碱性化合物会发生拖尾,这个原因是硅羟基的次级保留造成的,这里面的碱性化合物是指那些碱性物质?3.第二句话是化合物最好让流动相ph在化合物pka±2时候分析,酸性化和物是-2, 碱性化合物是+2(即在分子态分析)但是在实际应用中碱性化合物往往是在酸性的流动相下分析的?对于苯甲酸的pka是4.2但是现在测苯甲酸应用的乙酸铵流动相ph是6.67的貌似理论不能支持大部分的实践。请高手解答
关于三氧化二铝,看到很多不同的说法,有说是是共价化合物,有说是离子化合物,还有说是三氧化二铝是两种不同的结构,一种是共价化合物,一种是离子化合物。大家如何认为?
通常,我们所研究的都是具有手性中心的化合物。那具有手性轴、手性面的化合物有人研究吗?具有手性轴、手性面的化合物与具有手性中心的化合物有什么区别呢?他们的分离和分析方法类似吗?

于2009年5月28日,欧盟 (European Union, EU) 采纳了一项委员会决议(Commission Decision) 《2009/425/EC》,修正《76/769/EEC指令》中有关营销和使用「有机锡化合物」(organotin compounds) 的内容。这些化合物具有生物杀灭特性,并被广泛使用于多种消费品中。有机锡化合物会产生累积性的不良健康影响,即「经胸腺引起免疫毒性」(immunotoxicity via the thymus gland)。 新限制的实施是为了保护消费者,避免经由消费品接触这些化合物。新限制将禁止在物品或混合物中使用含锡重量超于 0.1% 的某些有机锡化合物。然而,各种化合物的生效日期均不同,要求概述如下。[B]背景[/B] 在某些情况下,用作生物杀灭剂的有机锡化合物,已在市场上被禁用。当中,「三取代有机锡化合物」(Tri-substituted organotin compound) 过往曾广泛被使用于船只防污漆,并可作为物品中的生物杀灭剂;而「双取代有机锡化合物」(di-substituted organotin compound) 则在消费品中被用作稳定剂或催化剂,尤其是「二丁基锡」(dibutyltin, DBT) 化合物和「二辛基锡」(dioctyltin, DOT) 化合物。[center][img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907121555_159672_1632583_3.jpg[/img][/center]如欲查阅决议全文,请浏览: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:138:0011:0013:EN:PDF
天然有机化合物结构鉴定方法摘自:汪茂田等, 化学工业出版社,200420 世纪下半叶,光谱学已成为有机化学的基础课程。30年代发展的紫外(UV)光谱和40年代的红外(IR)光谱为化学家提供了识别有机化合物生色基和官能团的有效方法。研究者可以采用极少量的样品,非破坏性的实验得到有关结构的信息。50年代发展起来的质谱(MS)方法进一步带来革命性的影响,MS实验可给出化合物的分子式,并且通过裂解方式提供分子的结构信息。 对有机结构化学影响最大的谱学方法当推核磁共振(NMR)。它对有机化学的影响是迅速的并且是震撼性的。近50年来,有机波谱学尤其是NMR技术的发展改革了天然产物结构鉴定的方法。波谱技术已成为探究大自然中分子内部秘密的最可靠、最有效的手段。今天波谱学已成为天然有机化学家不可或缺的工具。可以预期,即使在将来的千年岁月它们也一定是必不可少的。随着波谱技术的飞速发展,将会有更多的新技术为化学家所掌握,那时测定天然产物结构将会变得更加容易。 十几年前像HMBC、TOCSY等2DNMR等技术还是波谱学家刚开发的脉冲序列,可是在近几年市售的NMR波谱仪器上这些技术已成为常规方法了,这些技术已成为天然产物结构测定的强有力的工具。众所周知的吗啡(morphine))是1803年由Serturner分离得到的,直到1952年全合成成功才完成了结构确定,用了150年时间,番木鳖碱(Strychnine,士的宁)的结构确定用了半个多世纪(1891-1946),耗费了几代杰出化学家的心血,原因是当时确定结构的主要方法是湿法化学。而今天确定一个比较复杂的天然化合物的结构已变成研究生的科研训练课程,一般只需几个小时、几天、几周或几个月即可完成。这显然得益于波谱技术的发展和普及,同样重要的是一代又一代的天然有机化学家积累的波谱数据可供参考。严格地说,UV和IR属于光谱,MS不是光谱而是物质粒子的质量谱,NMR属于波谱。早年习惯称“四大光谱”,为了方便起见,本编中统称为波谱法。由于NMR技术在天然物结构测定中的重要地位,加之NMR技术解决天然产物结构问题的“多才多艺”,所以本编讨论的重点侧重于NMR方法。 用波谱法鉴定天然化合物结构需要的样品量。在进行天然物化学成分研究时或微量有机合成,一般分离出来的单体都是微量的,其量的范围通常在几mg至几十mg之间。当样品量大于10mg时,测定多种图谱已足够了。建议先测定NMR,因为测定了NMR的样品可以回收。 当只有几个mg样品且样品来之不易时,测定前需要有一个细致的方案。比如样品量约为5mg,取1~2mg 作为留样预防风险,其余3~4mg用于结构鉴定。根据研究者已获得的背景信息,如果该样品可能是已知化合物,测定1H、13CNMR和MS后,将样品回收再测定IR,与文献数据对照。按理,已知化合物鉴定的最方便的方法是找到对照样品和/或其IR图谱,可实际工作中对照品和对照IR图谱并非容易得到,文献中化合物的IR数据往往只报道几个最大吸收,这对鉴定一个化合物是不够的,因为用IR谱鉴定一个化合物要有图谱对照。如果可能是新化合物,尽可能不做燃烧分析而是采用高分辨MS来决定分子式和碎片离子的元素组成,因为测定MS所需样品量极微。完成各种先期必要的NMR图谱测定后,将样品暂时保留在样品管中,以备有疑问时进一步测定NMR,回收样品用来测定IR等。液体样品涂片测定IR后可用溶剂洗脱来回收,固体样品可从KBr 片中把样品回收。 作者曾用7mg二萜化合物进行酸催化重排反应(化学学报 1987,45,871),由甲醇得2.2mg无色结晶,显微熔点仪测定m.p 257~259℃,然后测定EIMS和1HNMR,回收NMR样品管中的样品,测定IR,再回收KBr 片中的样品。将最后的不足2mg样品进行燃烧分析(只能做一组数据)。 当用 2~3mg 样品测定多种NMR图谱时,花费较长的测试时间是不可避免的。建议尽可能使用磁场较高的仪器(如500MHz、600 MHz、800 MHz),因为磁场越高,灵敏度越高,可大大缩短测试时间,同时对化学位移非常相近的峰也能得到满意的分辨,更有利于图谱的解析。 当一个具有特殊意义的样品只有1 mg左右或者更少时,由于样品量甚微会使工作有一定难度。特别是微克级样品的结构测定,其工作本身就有一定难度,除了尽可能使用高磁场NMR谱仪外,使用微量探头(microprobe)或超低温微量探头测定NMR图谱是可取的。当然使用高分辨MS同样是不可少的。如果混合物中有一系列微克级成分需要鉴定,使用LC-NMR和[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]技术应是一个不错的选择。使用LC-NMR技术分离鉴定将在本章第三节的(四)部分介绍。 样品结构的背景信息 在进行结构鉴定之前,尽可能多地获得与样品化学结构有关的各种背景信息是非常必要的,信息量的多寡直接影响结构鉴定工作的速度。从各种植物中分离出来的成分可以说大部分是已知化合物,只要文献充足,这些已知物的结构鉴定一般都是比较快捷和容易的。对于一小部分新化合物,大多是已知骨架上的取代基不同和/或立体化学不同,文献数据对这些新化合物的鉴定起到非常重要的作用。研究中遇到全新骨架新化合物的几率比较低,即使是新骨架的新化合物,其部分结构单元往往也会在其它天然物结构中出现过,学习和积累天然物结构片段的波谱特征和特征数据,对于鉴定新化合物是很有用的,这也需要有文献数据的参考。所以详尽地查阅与所研究内容有关的文献是一件必要的工作。 在没有和/或缺乏背景信息的情况下,可以直接通过多种波谱技术测定化合物的结构(不包括X-ray四圆衍射法)。由于早期的文献受仪器功能的限制,有些数据没有经过可靠方法的证实(尽管结构没问题),波谱数据不一定完全可靠地归属。文献上通常把归属不清的数据用﹡和/或△表示,并注明了数据可以交换,立体化学未确定的基团用折线连接。由于波谱数据完全可靠的归属对于结构的确证和后人的参考有着重要的意义,所以近年来不断有国内外学者利用高场NMR波谱仪对一些复杂的天然产物进行1H和13NMR化学位移完全归属研究的文献发表,这类文章中的波谱数据很有参考价值。 原始文献和“二手文献”。原始文献是指研究者发表的学术论文,实验报告等,是最有参考价值的资料,作者把结构测定使用的方法、实验条件、分析讨论阐述的比较清楚,有的还有图谱。如果对原作者的结论有疑问,在网络发达的今天,可以很方便地和作者取得联系。所谓的“二手文献”指的是综述、专著、和手册等。综述,尤其是国外作者的综述,由于语言文字和信息来源广泛畅通的缘故更具有参考价值。这里并非贬低我国学者的综述文章,因为有一个不争的事实,那就是有些杂志在国内很难找到或根本找不到。质谱的应用
有做手性化合物分离的大佬吗?提供些手性化合物分离思路呗?感觉手性化合物分析起来不容易(要分离的化合物种类比较多,有多环类的,有氨基酸,多肽等)。
请问一下,我们ICP测无机化合物和有机化合物,何为无机化合物,何为有机化合物。能否具体举实例来说明一下呢?
合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。
公司新买了一台二手的AB API4000的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],在数据分析时发现最多能分析98个还是94个离子,也就是48个左右的化合物,多了就说超出范围,大家在测多种大混标时有遇见这个问题么?
请教下什么是脂肪族化合物/ 脂肪族醇類化合物他们有哪些化合物拜谢
合成化合物的结果是已知的,只要用谱和结构对照就可以知道化合物和预定的结构是否一致。对于植物中提取化合物的谱,首先应看是哪一类化合物,然后用已知的文献数据对照,看是否为已知物,如果文献中没有这个数据则继续测DEPT谱和二维谱,推出结构。对于一个全未知的化合物,除测核磁共振外,还要结合质谱、红外、紫外和元素分析,一步步推测结构。
在进行多种化合物同时分析时,经常会遇见两种化合物保留时间tR一致或相近成并肩峰,就是指前一色谱峰的峰尾还没有完全结束,后面的色谱峰峰前端已经出来,从而造成两峰完全重叠或成并肩峰。利用分离度R考察,一般R≤1时,认为两色谱峰有明显重叠。有时候是最开始就分离不开,有时候是原来能分开的,由于某些原因导致后来分不开。在未知样品检测中出现这种情况,就会导致我们无法对该色谱峰进行准确的定性和定量,那么到底有哪些原因会造成这种情况?遇到这种情况,该如何改善其分离效果,增加定性定量准确度?
下图是要分析的两个化合物。第一个化合物是第二个化合物的合成原料。现在要同时分析这两个化合物。这两个化合物遇水分解。目前只有GCFID和LCDAD设备,已经试过GC-FID但是峰型都很丑。有做过相关分析的大虾吗,求指教~[img]https://ng1.17img.cn/bbsfiles/images/2023/05/202305062129356141_9400_3135024_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/05/202305062129355391_1210_3135024_3.png[/img]
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
废水中汞和汞及其化合物是不是同一个项目?我们有汞的资质没有及其化合物的资质。能出报告吗?
大家好: 我对化合物简称弄不清楚,希望高手能赐教。 像三氟乙酸为什么是TFA,三氟乙酸乙酯是ETFA,三氟乙酸酐是TFAA等,像这种简称有一定的规律性吗?如果只告诉你简称,怎么来推化合物名称? 请各位多多帮忙!谢谢
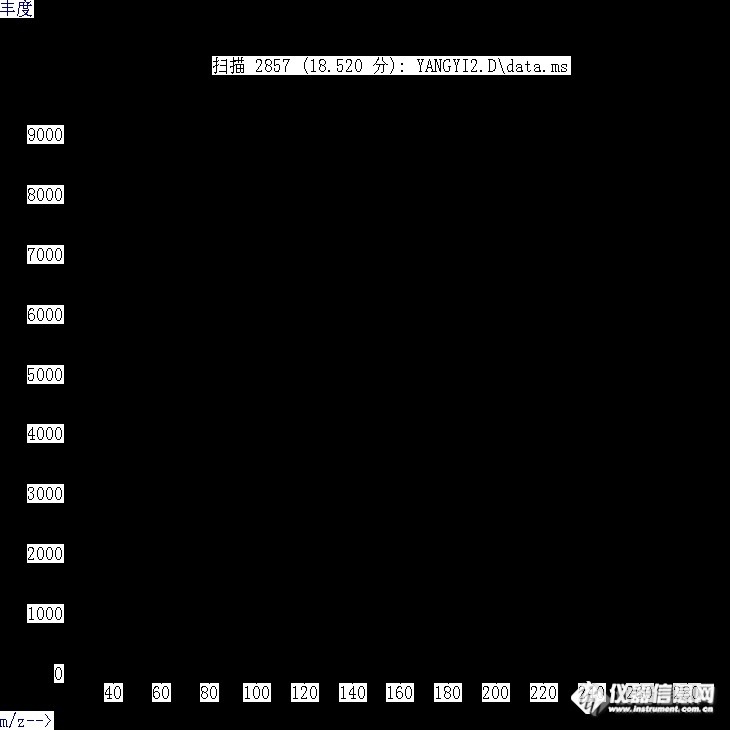
http://ng1.17img.cn/bbsfiles/images/2012/12/201212111503_411752_2657521_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212111503_411753_2657521_3.jpgGC-MS法测定了挥发油,结果有两个化合物的匹配度较低,我想鉴定,该怎么办。现将两个化合物的图片截图如上,请问该化合物是什么。
下列化合物中最难溶的化合物( ) 。 (A)AgNo3 (B)Agcl (C)Ag2cro4 (D)AgscN







 我要推广仪器
我要推广仪器
 下载APP
下载APP