糖酶的新近发展摘要:简要介绍了糖酶中淀粉酶、乳糖酶、纤维素酶作用机理和应用前景;详细介绍了α-葡萄糖苷酶的理化性质、作用机理、研究现状以及展望了其应用前景。关键词:糖酶;α-葡萄糖苷酶;研究现状;应用前景酶制剂作为一类绿色食品添加剂,用于改善食品品质和食品制造工艺,其应用已越来越普遍,品种也不断增多。其中,糖酶(carbohydrases)是一种应用相当广泛的酶类。它通过裂解多糖中将单糖结合在一起的化学键,使多糖降解成较小的分子;也能催化糖单位结构重排,形成新的糖类化合物,这类反应被称为转糖苷作用;此外,一些酯酶能作用于酯化的糖类,这类反应在改变果胶物质的功能性质上是很重要的。 本文大致介绍了糖酶中淀粉酶(amylase)、乳糖酶(lactase)、纤维素酶(cellulose)。主要介绍了α-葡萄糖苷酶(α-glucosidase),一种新型的糖苷酶。1.淀粉酶淀粉是由葡萄糖通过α-1 ,4糖苷键和α-1,6糖苷键连接而成的高分子化合物,而糖酶广泛分布于自然界,能裂解多糖中将单糖结合在一起的化学键,使多糖降解成为较小的分子,两者具体作用方式如下图所示。 淀粉工业中常用的糖酶有以下四类:(1)α-淀粉酶(液化酶,α-Amylase,α-1,4-葡聚糖葡萄糖水解酶,EC3.2.1.1),主要来自芽胞杆菌和曲霉,它是一种内切型淀粉酶,作用于淀粉是从淀粉分子的内部任意切开α-1,4-糖苷键,使淀粉分子迅速降解,失去粘性和碘的呈色反应,同时使降解物的还原力增强。(2)β-淀粉酶(β-Amylase,α-1,4-葡聚糖麦芽糖水解酶,EC3.2.1.2),在工业上应用的酶制剂主要来自植物(如大麦)和微生物(芽孢杆菌)。作为外切型淀粉酶,它作用于淀粉时从还原端以麦芽糖为单位切断α-1,4-糖苷键,但产物不是α型的麦芽糖,而是[font
请问,为什么糖酶解液用来跑氨基柱为什么不好跑呀?本来应该有最少6个峰的,可是只有一个峰,基线挺平的如图1,而用3K的超滤膜过滤蛋白后还是不行如图2,只是那个峰稍微高了一点,也不知道那个峰为什么会更高?其它峰怎么不见呢?[img=,690,480]https://ng1.17img.cn/bbsfiles/images/2019/10/201910101355036548_5671_1658948_3.png!w690x480.jpg[/img][img=,690,478]https://ng1.17img.cn/bbsfiles/images/2019/10/201910101355074842_3095_1658948_3.png!w690x478.jpg[/img]
根据文献等比例缩小体积,全部的反应均在96孔板上反应,先加入10μL底物(2%蔗糖溶液),后加入各浓度梯度的样品溶液20μL,反应5min后,加入蔗糖酶溶液10μL,反应20min后,加入无水乙醇20μL灭酶,灭酶后加入20μLDNS,最后用pbs稀释测定吸光度。(1)蔗糖酶用的是来自酵母菌的,我看文献用的都是鼠小肠里的(2)蔗糖酶一开始用的20U/mL,DNS测定后吸光度太大,且各个样品几乎没有抑制率,今天换2U/mL,样品和阿卡波糖均几乎没有抑制率。(3)究竟DNS测定这个是否存在问题,求老师指导。
在做饲用木聚糖酶酶活测定时,采用国标法结果很不稳定,同一样品隔几天测结果相差很大;标准曲线做了三次K值均呈下降趋势,相关系数倒是蛮好的。我怀疑是DNS试剂的问题,在整个配制过程中唯一不同的地方就是用滤纸过滤代替烧结玻璃过滤器过滤,请问影响大不?请各位帮忙分析下,谢谢!
http://file1.foodmate.net/file/upload/201202/17/16-21-31-65-410687.jpg关于对《木聚糖酶》行业标准征集意见的通知 附件: http://www.foodmate.net/member/fckeditor/editor/images/ext/zip.gif 木聚糖酶征求意见稿.zip
1.如题。GB/T23874-2009是检测饲料添加剂木聚糖酶活力的方法,现在我要检测猪饲料成品中的木聚糖酶活力,该采用什么方法?2.一般情况下猪饲料中木聚糖酶活力有多少?
谁有甘露聚糖酶的检测标准?
甘露聚糖酶如何检测?
请问:蔗糖酶滴定法的计算公式具体是怎样的?还有单位是什么?尽量精确一点,谢谢!!

乳制品的天然甜味帝斯曼乳糖酶健康是金,可以带来金钱和财富;健康是神圣的人权,是人们生存、发展、享乐的基本条件。随着健康意识的不断提升,人们开始用水果、低糖、无糖等健康产品来代替含糖量较高的食品和饮料,并希望通过各种方式减少糖分摄入。天然、简单、健康的低糖产品已成为一种主流的消费趋势。当今的食品行业日新月异,过去人们视为有益的成分现在也可能会损害消费者的健康。长期以来,生产商一直在不遗余力地减少甚至完全消除产品中的脂肪,然而最近他们开始将注意力转向糖分问题。http://ng1.17img.cn/bbsfiles/images/2015/08/201508292317_563484_1751239_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508292318_563486_1751239_3.png 减少糖分摄入被认为是预防超重、肥胖和龋齿的重要方式。此外,由于糖分摄入过多会增加罹患心脏病和糖尿病的风险,因此减少摄入还有利于身体健康。从摄入渠道来看,软饮料,谷类甜点,果汁、膳食、乳制品甜点,糖果4类食品中糖分含量位列前四。为减少糖分摄入,消费者需要口味和质量能够与糖相媲美的替代品;而生产商则需要在满足消费者日益增长的天然健康产品需求与保持产品口感之间寻求平衡。这为乳制品生产创新提供了众多机遇。讨论:帝斯曼乳糖酶是什么东东,和普通的乳糖酶有什么区别呢??谈谈您的看法,赢取积分
测定土样蔗糖酶时要在三角瓶中放置24小时,然后要6000rpm离心,这样就必须从三角瓶中移到离心管里,不用水冲洗的前提下有没有办法将三角瓶里的反应溶液倒干净,有没有必要倒干净,如果没法倒干净,直接将土样和测定的反应液放置在离心管里放置24小时,然后直接离心可以吗?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=67262]准确测定和表征β-葡聚糖酶活的方法与比较[/url]
GB/T23874-2009饲料添加剂木聚糖酶活力测定中4.6木聚糖溶液浓度为100mg/ml,有错误吗?因为其描述为:4.6木聚糖溶液浓度为100mg/ml:称取木聚糖(sigmaX0627)1.00g,加入0.32g氢氧化钠,再加入90ml水,磁力搅拌,同时缓慢加热,直至木聚糖完全溶解。然后停止加热,继续搅拌30min,加入0.5 ml冰乙酸。继续磁力搅拌,测定器pH,若PH为5.50,用乙酸-乙酸钠缓冲溶液定容至100ml。大家帮忙计算一下,国标上这样写的浓度是不是应该为10mg/ml啊?是不是国标出错了。另外sigmaX0627国内无现货,可以用别的代码的sigma药品啊?可以推荐下吗?急急急急急急
[align=left][font=宋体, SimSun][size=16px][b][font=宋体, SimSun]加工助剂在加工过程中发挥改善产品品质的功能,却在最终产品中没有残留,因为它们在加工过程中经常被销毁或移除。依据法规要求,这种物质不需要列在产品标签上,因此,使用加工助剂来代替化学添加剂为食品清洁标签提供了一种新途径。[/font][font=宋体, SimSun]酶制剂是一类最常用的加工助剂,在烘焙过程中,发挥改善功能的烘焙酶被高温烘烤所破坏,所以不必在标签上声明。因此,烘焙酶为烘焙食品带来了实现清洁标签的机会。[/font]木聚糖酶[i][/i]增加烘焙食品中的纤维含量[/b][/size][/font][/align][font=宋体, SimSun][size=16px]纤维有益于人体健康,所以很多面包师都试图增加烘焙产品中的纤维含量。然而,像麸皮这样的纤维,吸收了大量的水分,且吸收速度比面团中的其他成分较慢。[/size][/font][font=宋体, SimSun][size=16px][/size][/font][font=宋体, SimSun][size=16px]当纤维吸收水分后,面团变紧,导致操作不便。面团无法扩张,限制了酵母的作用,进而会导致面包质劣,且不能很好地成型。针对上述问题,木聚糖酶可切断大纤维链间的糖苷键[i][/i],以释放低分子量的糖和水。这有助于慢慢地软化面团,重新分配水分,增强气室延展性和稳定性,提高面团可操作性。[/size][/font][size=16px][/size][font=宋体, SimSun][size=15px][color=#48494d][/color][/size][/font]
用试剂盒方法(酶法)测定食品中葡聚糖含量摘要:描述了用试剂盒(酶法)测定食品中葡聚糖含量的方法前言: β-葡聚糖是禾谷类植物籽粒细胞壁中的多糖,是籽粒细胞壁的主要成分,化学名称为(1,3)(1,4) -β-D-葡聚糖,其含量以大麦和燕麦中较高其主要功能有:降低胆固醇,降血脂,调节血糖,提高免疫力,抗肿瘤和预防心血管疾病等。还有研究发现,它可以缓解和减轻肥胖症状。 β-葡聚糖测定方法包括酶测定法、荧光法、高效液相色谱法等,其中酶测定法因其反应专一性,测定准确可靠,而被采用为国际测定方法。酶法的方法过程如下:样品经水合和糊化,用地衣酶(β一葡聚糖酶)将样品中的β一葡聚糖酶解成β-葡基-寡聚糖。经调整体积和过滤分离后,用β一葡萄糖苷酶将这些可溶的B-葡基-寡聚糖水解成葡萄糖。葡萄糖用葡萄糖氧化酶氧化成葡萄糖酸和过氧化氢,后者用过氧化酶分解,以便在苯酚存在下,生色基4一氨基非那宗存在下形成适于比色分析的光吸收络合物,在紫外分光光度计下测定。 本文采用爱尔兰的Megazyme公司提供的测定(1,3)(1,4) -β-D-葡聚糖试剂盒,借鉴了EBC法3.11.1、AOAC法995.16,AACC法32-23方法,建立了测定相关制品中β-葡聚糖的测定方法。
微生物多糖包括某些细菌、真菌和蓝藻类产生的多糖,主要以三种形式存在:粘附在细胞表面上,分泌到培养基中,构成细胞的成分。微生物多糖,因其安全无毒、理化性质独特等优良性质而倍受关注。近几年,随着对微生物多糖研究的深入,世界上微生物多糖的产量和年增长量均在10%以上,而一些新型多糖年增长量在30%以上。到目前为止,已大量投产的微生物胞外多糖主要有黄原胶、热凝多糖、结冷胶、小核菌葡聚糖、短梗霉多糖等。微生物多糖具有植物多糖不具备的优良性质,它们生产周期短,不受季节、地域和病虫害条件限制,具有较强的市场竞争力和广阔的发展前景。随着对微生物多糖的结构和功能研究的不断深入,工业化的微生物多糖产品应用在各个领域,如美容养生的保健食品、工业染料的稳定剂、石油工业中的钻井泥浆处理剂、提高采油的注水稠化剂、意料中的代血浆、纺织造纸的上胶料、化妆品的拼料以及生物化学医药工业和实验室用的吸附剂、固定化酶或固定化细胞的载体等各个方面。微生物多糖的应用如此广泛,它的粘度如何呢?粘度是对流体内部摩擦的一种量度,是影响流体物理性质的一个重要参数。对于微生物多糖这种非牛顿流体来说,测定其粘度是鉴定其物理性质的一个重要方面。大部分非牛顿流体都是假塑性流体,特别是一些高分子溶液和悬浮液均具有剪切稀化的特性,假塑性流体的表观粘度随着剪切速率发生变化的范围很大,所以不能把它们作为牛顿流体来处理,必须对它的流动问题进行单独的测试。通常情况下,非牛顿流体的流变测量主要是在对流体施加一定剪切应力的条件下,通过跟踪流体对手里的响应值而获得。根据公式剪切应力Շ=kγn,k和n可以通过流变仪测出,但是流变仪价格昂贵,难以普及,因此可以通过测定不同剪切速率下的粘度值而计算出来。实验室采用美国BROOKFIELD公司的DV-S旋转粘度计测定流体的不同剪切速率下的粘度值,DV-S粘度计是BROOKFIELD最新研发的最经济的数字显示粘度计,采用全中文操作面板,操作简便,采用应力传感器,反应迅速,结合实验室仪器的使用可得到微生物多糖的粘度。
多糖在重水中溶解度是多少?
求助QB/T 4483-2013 木聚糖酶制剂QB/T 4481-2013 β-葡聚糖酶制剂
我现在想测多糖溶液的粘度,前期准备的是用乌式粘度计测量,但是近日发现实验室的仪器坏掉了,因此想改用流变仪测量溶液的粘度,我需要测一系列的,可不可以将每一样品冷冻干燥后再配制相同浓度溶液后再测其粘度,流变仪测粘度和乌式有什么不同,以前想从特性粘度与分子量对应起来,但流变仪可以求出特性粘度吗?
多糖的分析是一个大问题啊!和大家讨论一下吧,经综合各种文献我认为多糖结构分析内容:要搞清1. 多糖的单糖组成(种类、比例)2. 每个单糖残基的D-、L-构型,吡喃环式或呋喃环式3. 羟基被取代情况(糖苷键的位置)4. 糖苷键及构型(α、β异头异构体)5. 重复单元方法:1、单糖组成:(对照品:葡萄糖、岩藻糖、半乳糖、甘露糖、木糖、阿拉伯糖、鼠李糖)a:水解: 纸层析薄层层析气相色谱(糖氰乙酸酯衍生物、糖醇乙酸酯)液相色谱(ZORBAX-NH2、HRC-NH2、RID)首选气相,灵敏度高,液相RSD、ELSD灵敏度低b:TFA酸解:气相色谱(乙酰化物)c:甲醇解:气相色谱(三甲基硅醚)2:高碘酸钠氧化和Smith降解a:每摩尔己糖基的高碘酸消耗量、甲酸释放量。(目的:判断可氧化糖基与不可氧化糖基之比例)b:Smith降解完全水解,气相分析,如有葡萄糖(表示有1-3键糖基)、甘油(有1-6或1-2糖基)、甲酸(有1-6糖基)Smith降解部分水解,说明主干糖苷键类型。3:甲基化分析(Hakrmor法)-支链分布多糖—甲基化—水解—还原得甲基化单糖醇—乙酰化得糖醇衍生物—GC-MS检测。 对照品 2,3,4,6-四甲基葡萄糖 糖苷键类型 1—2,4,6- 三甲基葡萄糖 1—32,3,4-三甲基葡萄糖 1—62,4-二甲基葡萄糖 1—3,6 4:IR图谱解析a:吡喃环式或呋喃环式α、β异头异构体5:1HNMR及13CNMR解析(构型)6:纯度检查:a: 紫外吸收光谱(280、260)b:电泳(琼脂糖电泳、聚丙烯酰胺凝胶、醋酸纤维素薄膜)c:薄层色谱(多糖不水解)7:X射线衍射,立体构型。好多啊!想和大家讨论讨论多糖的HPLC分析,我们试验室用的液相是C18柱,紫外检测器,做多糖含量及纯度检测,这样的装备够不够用呀?是不是做前必须衍生化或有其它方法,如用示差折射仪作检测器,是不是不需衍生化?多糖的HPLC分析,用得较多用HPGPC测分子量及分子量分布。一般纯多糖紫外吸收较弱,多用RID或ELSD。至于含量测定多用硫酸蒽酮比色或苯酚硫酸法。http://img.dxycdn.com/images_new/smiles/smile_angry.gif
本人最近测定枸杞多糖含量时,所用测定方法均为2005年版药典枸杞项下,结果出现如下问题:测定枸杞提取物(注:厂家未提供提取方法)枸杞多糖含量时,直接称样水溶解然后测定,结果所测提取物含量高达95%;按药典方法进行前处理,测出值也高达70%;而厂家提供的是50%。另外,我们公司有一种产品,45度白酒,其中添加了葛根黄酮提取物和这种枸杞提取物,结果所测枸杞多糖含量也与理论添加量有很大差别。请问:像我这种情况,在测定枸杞多糖时,需要像药典中那样前处理吗?有哪些更好的方法?多糖测定时葛根黄酮会不会影响?测定提取物和测定白酒中多糖含量时方法是否可以一致?不行的话又分别该怎样测定? 恳请问各位专家朋友指点迷津!谢谢![/color][/color][/color]
[color=#444444]之前用水提醇沉法提了黄芪多糖,课题要求检测其水解后单糖组成及比例,目前做到单糖组成这一步。[/color][color=#444444]水解用的是三氟乙酸,要求完全水解,对比条件中最剧烈的条件是5M三氟乙酸,100度水解10h,但是就算是这个条件,我进[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]时依然检测不到文献中说到的理论应有单糖,甚至也检测不到多糖,请各位帮忙找找原因。[/color][color=#444444]说说之前做的,水解后我有氮气吹干,保存于-80℃冰箱,几个条件都考察完后一起冻干。[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]样品处理:75%乙腈和25%乙醇溶解水解后样品,离心,吸取上清。[/color][color=#444444]所用色谱柱:HILIC柱。[/color][color=#444444]后来也打了单质谱,依旧检测不出所需要的物质。[/color][color=#444444]不知道是哪一步出错了,请大家帮忙,多谢![/color]
[color=#fe2419][size=2]维权声明:本文为19861005原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[b][size=3]多糖提取分离的基本流程[/size][/b][/size][/color][font=KaiTi_GB2312][size=4] 长期以来,天然药物化学与药理学研究工作侧重于脂溶性化学成分,如生物碱,苷类等,而对水溶性多糖类有效成分重视不够,在天然药物活性成分研究中往往将多糖作为杂质除去。近年来,国内外对多糖的研究比较活跃,其作为生物效应调节剂,主要具有抗肿瘤,抗炎,抗凝血,抗病毒,降血糖,降血脂等活性。[/size][size=4][/size][size=4] 今天我就和大家探讨一下多糖的提取分离方法。[/size][size=4][/size][color=#fe2419][size=4][b](一)提取[/b][/size][/color][size=4][/size][size=4] 水提得到的粗多糖水溶液,加入[/size][size=4]95%[/size][size=4]乙醇使含醇量达[/size][size=4]80%[/size][size=4],[/size][size=4]4[/size][size=4]摄氏度静置过夜,多糖析出。减压抽滤,用[/size][size=4]95%[/size][size=4]乙醇洗至上清液无色,挥干醇。[/size][size=4][/size][color=#fe2419][size=4][b](二)除蛋白[/b][/size][/color][size=4][/size][size=4] 这是至关重要的一步,由于蛋白是大分子,它的存在会干扰多糖的纯化。一般选用[/size][size=4]sevage[/size][size=4]法萃取或离心,多糖浓度为[/size][size=4]1mg/ml, [/size][size=4]氯仿[/size][size=4]-[/size][size=4]正丁醇比例通常在[/size][size=4]3:1~5:1[/size][/font][font=KaiTi_GB2312][size=4]之间,变性的蛋白质一般在两相交界处产生一条白色的带。建议进行小试,确定最佳比例,同时确定除蛋白的次数,如果含量较多建议八次以上,含量较少的话四到五次即可。[b][color=#fe2419](三)检测[/color][/b][/size][size=4][/size][size=4] 除蛋白前后用紫外分光光度计检测[/size][size=4]280nm[/size][size=4](蛋白质)和[/size][size=4]260nm[/size][size=4](核酸)下的吸收值,多糖浓度为[/size][size=4]0.4mg/ml[/size][size=4]对照验证除蛋白的效果。需注意的是氯仿的比例不宜太大,否则可能会破坏多糖。萃取时产生的乳化现象,可用超声除去。除蛋白后的多糖水溶液会有很浓的有机试剂味道,可以用悬蒸除去。[/size][size=4][/size][b][color=#fe2419][size=4]([/size][size=4]四[/size][size=4])[/size][size=4]离子交换纤维素[/size][size=4](DEAE-[/size][size=4]纤维素[/size][size=4])[/size][size=4]进行初步分离[/size][size=4][/size][/color][/b][size=4] [/size][size=4]一般选用[/size][size=4]DE-52[/size][size=4]型离子交换纤维素[/size][size=4][/size][b][color=#0162f4][size=4][/size][size=4]柱的预处理[/size][size=4][/size][/color][/b][size=4] [/size][size=4]加蒸馏水浸泡过夜,期间换几次水,每次除去细小颗粒,,用[/size][size=4]0.5mol/LHCl[/size][size=4]溶液浸泡[/size][size=4]1~2h, [/size][size=4]蒸馏水漂洗至中性;再用[/size][size=4]0.5mol/LNaOH[/size][size=4]溶液浸泡[/size][size=4]1~2h, [/size][size=4]蒸馏水漂洗至中性。[/size][size=4][/size][b][color=#2690fe][size=4][/size][size=4]洗脱[/size][size=4][/size][/color][/b][size=4] [/size][size=4]样品浓度在[/size][size=4]20mg/ml[/size][size=4]左右。先用水洗脱,再用[/size][size=4]NaCl[/size][size=4]梯度洗脱,小试时的梯度应密集一些,[/size][size=4]0.05, 0.1, 0.3, 0.5mol/L[/size][size=4]洗脱,每瓶[/size][size=4]8ml, 10min[/size][size=4]左右一瓶即可。根据紫外检测的结果,再进行梯度的修改。[/size][size=4][/size][color=#0162f4][b][size=4][/size][size=4]合并流分[/size][/b][size=4][/size][/color][size=4] [/size][size=4]硫酸[/size][size=4]-[/size][size=4]蒽酮法结合紫外检测[/size][size=4][/size][b][color=#fe9a95][size=4]([/size][size=4]1[/size][size=4])硫酸[/size][size=4]-[/size][size=4]蒽酮试剂的配制[/size][size=4][/size][/color][/b][size=4]称取[/size][size=4]0. 1g[/size][size=4]蒽酮至容量瓶中,加少量浓硫酸,搅拌使其溶解,加浓硫酸至[/size][size=4]50ml[/size][size=4]摇匀,得[/size][size=4]0.2%[/size][size=4]的硫酸[/size][size=4]-[/size][size=4]蒽酮试剂,尽量现配先用,也可放[/size][size=4]4[/size][size=4]摄氏度冷藏备用,若颜色变绿,则说明已变质不能使用。[/size][size=4][/size][color=#fe9a95][b][size=4]([/size][size=4]2[/size][size=4])方法[/size][/b][/color][size=4][/size][size=4]馏分隔管检测,每管取[/size][size=4]0.5ml[/size][size=4],在冰浴中加入[/size][size=4]2ml[/size][size=4]硫酸[/size][size=4]-[/size][size=4]蒽酮试剂,然后在沸水中煮[/size][size=4]10~15min, [/size][size=4]自来水冷却,放至室温。[/size][size=4][/size][color=#fe9a95][b][size=4]([/size][size=4]3[/size][size=4])紫外检测[/size][/b][/color][size=4][/size][size=4] [/size][size=4]主要看[/size][size=4]610nm[/size][size=4]出吸收值,合并馏分。[/size][size=4][/size][b][color=#f10b00][size=4](五)凝胶[/size][size=4]G[/size][size=4]进一步纯化[/size][/color][/b][size=4] [/size][size=4] [/size][size=4]通常采用[/size][size=4]G-100[/size][size=4]或[/size][size=4]G-200[/size][size=4]不断进行纯化[/size][size=4] [/size][b][color=#f10b00][size=4](六)纯度检测[/size][size=4][/size][/color][/b][size=4] [/size][size=4]采用高效液相凝胶柱进行检测[/size][size=4][/size][size=4][b]总结[/b] 以上就是本人在多糖提取分离过程中的一些步骤和小心得,愿与大家分享,互相学习,互相进步,共同为加快多糖研究的进展推波助澜~[img]http://simg.instrument.com.cn/bbs/images/brow/em09502.gif[/img][/size][/font][size=4][font=Times New Roman][/font][/size]
本人最近测定枸杞多糖含量时,所用测定方法均为2005年版药典枸杞项下,结果出现如下问题:测定枸杞提取物(注:厂家未提供提取方法)枸杞多糖含量时,直接称样水溶解然后测定,结果所测提取物含量高达95%;按药典方法进行前处理,测出值也高达70%;而厂家提供的是50%。另外,我们公司有一种产品,45度白酒,其中添加了葛根黄酮提取物和这种枸杞提取物,结果所测枸杞多糖含量也与理论添加量有很大差别。请问:像我这种情况,在测定枸杞多糖时,需要像药典中那样前处理吗?有哪些更好的方法?多糖测定时葛根黄酮会不会影响?测定提取物和测定白酒中多糖含量时方法是否可以一致?不行的话又分别该怎样测定? 恳请问各位专家朋友指点迷津!谢谢!
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网[em0805]
用《保健食品功效成分检测方法》的方法测口服液的粗多糖(按葡聚糖计算),得到的结果比理论值低几倍,且不稳定,个人认为出问题的主要是铜试剂沉淀葡聚糖这一步,求有遇到类似问题的朋友指导要点…
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网
因为课题需要,现需要将蛋白质和多糖的混合液分开,并且需要把各种蛋白质和各种多糖都分开,但是我不知道具体是哪些蛋白质和多糖,请问各位有什么可行的方法没得?
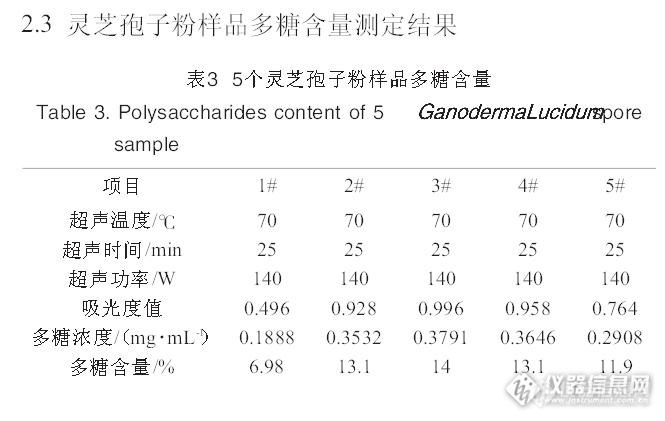
中国药典2010年版一部关于灵芝药材的多糖含量测定问题偶然的机会,有个样品是灵芝药材,做全检,其中含量测定是做灵芝的多糖含量.多糖含量做的多了,但都是保健食品的检验项目,还有做枸杞子的多糖.这个灵芝的多糖没做过.按常理这个灵芝多糖该很顺利就做出来的.可是事与愿违.按药典方法做出来的结果大跌眼镜,多糖含量结果是0.之后就是找原因,先找个人操作问题,步骤没错,顺序也没错,试剂也没加错.那操作排除了.又去药店买了整个的灵芝回来作参照,结果也是大跌眼镜,一样测出来的结果是0。问题来了。细细观察过程,原来在醇沉的那步骤里有异常,送检的灵芝和买回来的灵芝都没有沉淀产生,溶液还是透明的。难道是多糖量少的原因,于是又按药典的量称了1mg葡聚糖对照品用2ml水溶解后,再醇沉,是阳性反应啊,有乳白色的浑浊产生(沉淀)。http://ng1.17img.cn/bbsfiles/images/2013/10/201310171824_471607_1621232_3.jpg后来又拿了黄芪药材作参照(黄芪含有大量的黄芪多糖),在醇沉的那步骤里也是有乳白色的浑浊产生啊(沉淀)。到这里,该推断出问题该出在样品上了。后来又查了一下文献,有文献说到灵芝饱子粉含有的多糖含量大约为10%左右。http://ng1.17img.cn/bbsfiles/images/2013/10/201310171826_471610_1621232_3.jpg最后,我又把送检灵芝和买来的灵芝做了薄层鉴别,薄层是与对照药材一样有荧光斑点的哦。到这里,想到一个原因,现在灵芝饱子粉热销,很值钱,很多的种植户都是待灵芝开了后收集了饱子粉才会采摘整个的灵芝子实体,图经济效应最大化。所以以前我们还可以买到子实体(带有泥灰一样的),现在的买到子实体都是非常的干净,干净到发亮反光(哈哈,有点夸张吧)。我是这样想的,为什么这次送检的灵芝和药店买回来的灵芝做不出多糖呢,问题在于饱子粉上,饱子粉含多糖达到10%,而子实体基本是木头(纤维),子实体的多糖该很低的,子实体开过收集了饱子粉以后,剩下的没有孢子的子实体的多糖数值肯定没多少了。做不出来也难怪了。所以,在修定药材标准时,特别是整株植物药材,由几个不同的部位组成的,有花有果有茎有叶有根的,就更复杂了。一定要弄清楚目标物的来源,现有的商人都是追逐利润最大化的。

灵芝子实体粗多糖提取及分析灵芝多糖具提高免疫力、抗氧化、抗肿瘤、安神、降血糖、消除放化疗副反应、除胃热、保肝解毒等功效。灵芝多糖是灵芝中最有效的成分之一,因此,也特别受到医药科技工作者的重视,研究报道也最多。现知灵芝多糖有广泛的药理活性,能提高机体免疫力,提高机体耐缺氧能力,消除自由基,抑制肿瘤、抗辐射,提高肝脏、骨髓、血液合成DNA、RNA、蛋白质能力,延长寿命,灵芝多糖还具有刺激宿主非特异性抗性、免疫特异反应以及抑制移植肿瘤生理活性的特性。材料实验所用材料购于市场,经鉴定为灵芝。前处理取灵芝子实体500g剪碎,用75%的乙醇回流脱酯2小时,反复三次,离心,上清液用旋转蒸发仪蒸除乙醇,得灵芝浸膏。提取灵芝残渣分别用pH=2的盐酸溶液、6%的尿素溶液、3%的三氯醋酸溶液室温提取24小时,透析,离心,上清液加入4倍85%乙醇醇析,得粗多糖FS。分级灵芝子实体经脱脂,分别用上述三种方法提取得粗多糖FS,对其中的FS水溶粗多糖进行乙醇分级。FS配成5%多糖溶液,搅拌滴加乙醇,使乙醇的浓度依次达到30%、50%、70%离心所得沉淀依次为FS-A、FS-B、FS-C。脱蛋白5%糖溶液,用链蛋白酶,按酶:蛋白=l:50取蛋白酶,加入糖溶液中,加少量二甲苯防腐,1%NaCI作激活剂,37度,保温24小时,按多糖液总体积1/4加入Sevgae试剂(氯仿:正丁醇=4[/size







 我要推广仪器
我要推广仪器
 下载APP
下载APP