2689高效液相色谱常见故障的断定及解决
最近看了一篇文献,作者在XRD图中说(100)面比标准卡片的峰强,从而晶体沿着此面取向生长;然后,作者又提供了样品局部的SAED和HRTEM,通过分析,作者说晶体(纳米棒)沿着【010】生长。 我想请教的是:二维HRTEM(纳米棒局部)很明显可以断定晶体生长方向,并且和SAED相互印证,是不是XRD就不需要了?并且作者说的取向生长方向不一样,怎么解释????? 谢谢
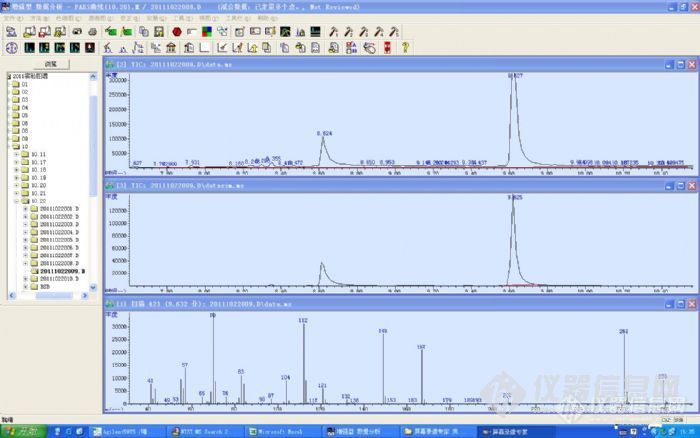
仪器型号:安捷伦6890-5975柱子:30mX0.25mmX0.25um采用全扫描和离子扫描模式DNOP标液的出峰时间是9.571,样品的是9.627标液特征离子149 207 281 ,而样品中却出现了很多峰,如:149 167 261 279且比例也不匹配纠结不能断定是不是DNOP,请各位大侠帮助。这个是标液的图谱:http://ng1.17img.cn/bbsfiles/images/2011/10/201110251649_326268_2281619_3.jpg下面的是样品的图谱http://ng1.17img.cn/bbsfiles/images/2011/10/201110251650_326271_2281619_3.jpg
色谱高效液相色谱常见故障的断定及解决[color=red]【由于该附件或图片违规,已被版主删除】[/color][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=10334]111[/url]
请问,测得的XRD谱图的衍射角的误差在多大范围内才能断定是标准卡片的结构。我们测得是一种矿物,纯度在百分之八十五左右。谢谢!
[b]‘有奖问答’选择题’顾客小王买了[font=宋体][color=#6D6D6D]治疗腰肌劳损的频谱治疗仪,用后腰肌劳损大大减轻,但却患上了头疼症,[b]小王私下[/b]询问了这种治疗仪的其他用户,很多人都有类似反应。[b]小王收集完口供后[/b]向厂家索赔。生产厂家对此十分重视,专门找第三方权威机构作了鉴定;结论是:目前科学技术无法断定治疗仪与头疼之间的关系。以下哪种观点正确( )A.本着公平,负责的原则,厂家应予适当赔偿B.因出现不良反应的用户众多,应将争议搁置,待科技发展到能够作出明确结论时再处理C.该治疗仪的功能是治疗腰肌劳损,该功能完全具备,至于其他副作用是治疗中不可避免的,该厂可不负责任D.由于治疗仪投入流通时的科学技术水平不能发现缺陷的存在,厂家不承担赔偿责任[/color][/font][/b]
高效液相色谱常见故障的断定及解决诊状 可能的原因 解 决 方 法 (一)保留时间变化 1.柱温变化 柱恒温,必要时需配置恒温箱 2.等度与梯度间未能充分平衡 至少用10倍柱体积的流动相平衡柱 3.缓冲液容量不够 用25mmol/L的缓冲液 4.柱污染 每天冲洗柱 5.柱内条件变化 稳定进样条件,调节流动相 6.柱快达到寿命 采用保护柱 (二)保留时间缩短 1.流速增加 检查泵,重新设定流速 2.样品超载 降低样品量 3.键合相流失 流动相PH值保持在3~7.5检查柱的方向 4.流动相组成变化 防止流动相蒸发或沉淀 5.温度增加 柱恒温 (三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡 2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱 3.键合相流失 同前(二)3 4.流动相组成变化 同前(二)4 5.温度降低 同前(二)5 (四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15% 2.样品溶剂过强 采用较弱的样品溶剂 3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件 4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品 5.进样器损坏 更换进样器转子 (五)鬼峰 1.进样阀残余峰 每次用后用强溶剂清洗阀,改进阀和样品的清洗 2.样品中未知物 处理样品 3.柱未平衡 重新平衡柱,用流动相作样品溶剂 (尤其是离子对色谱) 4.三氟乙酸(TFA)氧化(肽谱) 每天新配,用抗氧化剂 5.水污染(反相) 通过变化平衡时间检查水质量,用HPLC级的水 (六) 基线噪声 1.气泡(尖锐峰) 流动相脱气,加柱后背压 2.污染(随机噪声) 清洗柱,净化样品,用HPLC级试剂 3.检测器灯连续噪声 更换氘灯 4.电干扰(偶然噪声) 采用稳压电源,检查干扰的来源(如水浴等) 5.检测器中有气泡 流动相脱气,加柱后背压 (七)峰拖尾 1.柱超载 降低样品量,增加柱直径采用较高容量的固定相 2.峰干扰 清洁样品,调整流动相 3.硅羟基作用 加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品 4.同前(四)4 同前(四)4 5.同前(四)3 5.同前(四)3 6.死体积或柱外体积过大 连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管 7.柱效下降 用较低腐蚀条件,更换柱,采用保护柱 (八)峰展宽 1.进样体积过大 同(四)1 2.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散 3.数据系统采样速率太慢 设定速率应是每峰大于10点 4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10% 5.流动相粘度过高 增加柱温,采用低粘度流动相 6.检测池体积过大 用小体积池,卸下热交换器 7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱 8.柱外体积过大 将连接管径和连接管长度降至最小 9.样品过载 进小浓度小体积样品
诊状 可能的原因 解 决 方 法 (一)保留时间变化 1.柱温变化 柱恒温,必要时需配置恒温箱2.等度与梯度间未能充分平衡 至少用10倍柱体积的流动相平衡柱3.缓冲液容量不够 用 25mmol/L的缓冲液4.柱污染 每天冲洗柱5.柱内条件变化 稳定进样条件,调节流动相6.柱快达到寿命 采用保护柱(二)保留时间缩短 1.流速增加 检查泵,重新设定流速2.样品超载 降低样品量3.键合相流失 流动相PH值保持在3~7.5检查柱的方向4.流动相组成变化 防止流动相蒸发或沉淀5.温度增加 柱恒温(三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱3.键合相流失 同前(二)34.流动相组成变化 同前(二)45.温度降低 同前(二)5(四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15%2.样品溶剂过强 采用较弱的样品溶剂3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品5.进样器损坏 更换进样器转子
在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]的选择离子检测中,我们选择两组离子对来定性,请问选择其中那一对来定量呢?是丰度较大的么?
25mmol/L的缓冲液4.柱污染 每天冲洗柱5.柱内条件变化 稳定进样条件,调节流动相6.柱快达到寿命 采用保护柱(二)保留时间缩短 1.流速增加 检查泵,重新设定流速2.样品超载 降低样品量3.键合相流失 流动相PH值保持在3~7.5检查柱的方向4.流动相组成变化 防止流动相蒸发或沉淀5.温度增加 柱恒温(三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱3.键合相流失 同前(二)34.流动相组成变化 同前(二)45.温度降低 同前(二)5(四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15%2.样品溶剂过强 采用较弱的样品溶剂3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品5.进样器损坏 更换进样器转子(五)鬼峰1.进样阀残余峰 每次用后用强溶剂清洗阀,改进阀和样品的清洗2.样品中未知物 处理样品3.柱未平衡 重新平衡柱,用流动相作样品溶剂 (尤其是离子对色谱)4.三氟乙酸(TFA)氧化(肽谱) 每天新配,用抗氧化剂5.水污染(反相) 通过变化平衡时间检查水质量,用HPLC级的水(六) 基线噪声 1.气泡(尖锐峰) 流动相脱气,加柱后背压2.污染(随机噪声) 清洗柱,净化样品,用HPLC级试剂3.检测器灯连续噪声 更换氘灯4.电干扰(偶然噪声) 采用稳压电源,检查干扰的来源(如水浴等)5.检测器中有气泡 流动相脱气,加柱后背压(七)峰拖尾 1.柱超载 降低样品量,增加柱直径采用较高容量的固定相2.峰干扰 清洁样品,调整流动相3.硅羟基作用 加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品4.同前(四)4 同前(四)45.同前(四)3 5.同前(四)36.死体积或柱外体积过大 连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管7.柱效下降 用较低腐蚀条件,更换柱,采用保护柱(八)峰展宽 1.进样体积过大 同(四)12.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散3.数据系统采样速率太慢 设定速率应是每峰大于10点4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10%5.流动相粘度过高 增加柱温,采用低粘度流动相6.检测池体积过大 用小体积池,卸下热交换器7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱8.柱外体积过大 将连接管径和连接管长度降至最小9.样品过载 进小浓度小体积样品
诊状 可能的原因 解 决 方 法 (一)保留时间变化 1.柱温变化 柱恒温,必要时需配置恒温箱2.等度与梯度间未能充分平衡 至少用10倍柱体积的流动相平衡柱3.缓冲液容量不够 用〉25mmol/L的缓冲液4.柱污染 每天冲洗柱5.柱内条件变化 稳定进样条件,调节流动相6.柱快达到寿命 采用保护柱(二)保留时间缩短 1.流速增加 检查泵,重新设定流速2.样品超载 降低样品量3.键合相流失 流动相PH值保持在3~7.5检查柱的方向4.流动相组成变化 防止流动相蒸发或沉淀5.温度增加 柱恒温(三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱3.键合相流失 同前(二)34.流动相组成变化 同前(二)45.温度降低 同前(二)5(四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15%2.样品溶剂过强 采用较弱的样品溶剂3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品5.进样器损坏 更换进样器转子(五)鬼峰1.进样阀残余峰 每次用后用强溶剂清洗阀,改进阀和样品的清洗2.样品中未知物 处理样品3.柱未平衡 重新平衡柱,用流动相作样品溶剂 (尤其是离子对色谱)4.三氟乙酸(TFA)氧化(肽谱) 每天新配,用抗氧化剂5.水污染(反相) 通过变化平衡时间检查水质量,用HPLC级的水(六) 基线噪声 1.气泡(尖锐峰) 流动相脱气,加柱后背压2.污染(随机噪声) 清洗柱,净化样品,用HPLC级试剂3.检测器灯连续噪声 更换氘灯4.电干扰(偶然噪声) 采用稳压电源,检查干扰的来源(如水浴等)5.检测器中有气泡 流动相脱气,加柱后背压(七)峰拖尾 1.柱超载 降低样品量,增加柱直径采用较高容量的固定相2.峰干扰 清洁样品,调整流动相3.硅羟基作用 加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品4.同前(四)4 同前(四)45.同前(四)3 5.同前(四)36.死体积或柱外体积过大 连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管7.柱效下降 用较低腐蚀条件,更换柱,采用保护柱(八)峰展宽 1.进样体积过大 同(四)12.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散3.数据系统采样速率太慢 设定速率应是每峰大于10点4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10%5.流动相粘度过高 增加柱温,采用低粘度流动相6.检测池体积过大 用小体积池,卸下热交换器7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱8.柱外体积过大 将连接管径和连接管长度降至最小9.样品过载 进小浓度小体积样品
诊状可能的原因解决方法 (一)保留时间变化 1.柱温变化柱恒温,必要时需配置恒温箱 2.等度与梯度间未能充分平衡至少用10倍柱体积的流动相平衡柱 3.缓冲液容量不够用》25mmol/L的缓冲液 4.柱污染每天冲洗柱 5.柱内条件变化稳定进样条件,调节流动相 6.柱快达到寿命采用保护柱 (二)保留时间缩短 1.流速增加检查泵,重新设定流速 2.样品超载降低样品量 3.键合相流失流动相PH值保持在3~7.5检查柱的方向 4.流动相组成变化防止流动相蒸发或沉淀 5.温度增加柱恒温 (三)保留时间延长 1.流速下降管路泄漏,更换泵密封圈,排除泵内气泡 2.硅胶柱上活性点变化用流动相改性剂,如加三乙胺,或采用碱至钝化柱 3.键合相流失同前(二)3 4.流动相组成变化同前(二)4 5.温度降低同前(二)5 (四)出现肩峰或分叉 1.样品体积过大用流动相配样,总的样品体积小于第一峰的15% 2.样品溶剂过强采用较弱的样品溶剂 3.柱塌陷或形成短路通道更换色谱柱,采用较弱腐蚀性条件 4.柱内烧结不锈钢失效更换烧结不锈钢,加在线过滤器,过滤样品 5.进样器损坏更换进样器转子 (五)鬼峰 1.进样阀残余峰每次用后用强溶剂清洗阀,改进阀和样品的清洗 2.样品中未知物处理样品 3.柱未平衡重新平衡柱,用流动相作样品溶剂(尤其是离子对色谱)4.三氟乙酸(TFA)氧化(肽谱)每天新配,用抗氧化剂 5.水污染(反相)通过变化平衡时间检查水质量,用HPLC级的水 (六)基线噪声 1.气泡(尖锐峰)流动相脱气,加柱后背压 2.污染(随机噪声)清洗柱,净化样品,用HPLC级试剂 3.检测器灯连续噪声更换氘灯 4.电干扰(偶然噪声)采用稳压电源,检查干扰的来源(如水浴等) 5.检测器中有气泡流动相脱气,加柱后背压 (七)峰拖尾 1.柱超载降低样品量,增加柱直径采用较高容量的固定相 2.峰干扰清洁样品,调整流动相 3.硅羟基作用加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品 4.同前(四)4同前(四)4 5.同前(四)35.同前(四)3 6.死体积或柱外体积过大连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管 7.柱效下降用较低腐蚀条件,更换柱,采用保护柱 (八)峰展宽 1.进样体积过大同(四)1 2.在进样阀中造成峰扩展进样前后排出气泡以降低扩散 3.数据系统采样速率太慢设定速率应是每峰大于10点 4.检测器时间常数过大设定时间常数为感兴趣第一峰半宽的10% 5.流动相粘度过高增加柱温,采用低粘度流动相 6.检测池体积过大用小体积池,卸下热交换器 7.保留时间过长等度洗脱时增加溶剂含量也可用梯度洗脱 8.柱外体积过大将连接管径和连接管长度降至最小 9.样品过载进小浓度小体积样品
诊状 可能的原因 解 决 方 法 (一)保留时间变化 1.柱温变化 柱恒温,必要时需配置恒温箱 2.等度与梯度间未能充分平衡 至少用10倍柱体积的流动相平衡柱 3.缓冲液容量不够 用25mmol/L的缓冲液 4.柱污染 每天冲洗柱 5.柱内条件变化 稳定进样条件,调节流动相 6.柱快达到寿命 采用保护柱 (二)保留时间缩短 1.流速增加 检查泵,重新设定流速 2.样品超载 降低样品量 3.键合相流失 流动相PH值保持在3~7.5检查柱的方向 4.流动相组成变化 防止流动相蒸发或沉淀 5.温度增加 柱恒温 (三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡 2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱 3.键合相流失 同前(二)3 4.流动相组成变化 同前(二)4 5.温度降低 同前(二)5 (四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15% 2.样品溶剂过强 采用较弱的样品溶剂 3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件 4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品 5.进样器损坏 更换进样器转子 (五)鬼峰 1.进样阀残余峰 每次用后用强溶剂清洗阀,改进阀和样品的清洗 2.样品中未知物 处理样品 3.柱未平衡 重新平衡柱,用流动相作样品溶剂 (尤其是离子对色谱) 4.三氟乙酸(TFA)氧化(肽谱) 每天新配,用抗氧化剂 5.水污染(反相) 通过变化平衡时间检查水质量,用HPLC级的水 (六) 基线噪声 1.气泡(尖锐峰) 流动相脱气,加柱后背压 2.污染(随机噪声) 清洗柱,净化样品,用HPLC级试剂 3.检测器灯连续噪声 更换氘灯 4.电干扰(偶然噪声) 采用稳压电源,检查干扰的来源(如水浴等) 5.检测器中有气泡 流动相脱气,加柱后背压 (七)峰拖尾 1.柱超载 降低样品量,增加柱直径采用较高容量的固定相 2.峰干扰 清洁样品,调整流动相 3.硅羟基作用 加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品 4.同前(四)4 同前(四)4 5.同前(四)3 5.同前(四)3 6.死体积或柱外体积过大 连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管 7.柱效下降 用较低腐蚀条件,更换柱,采用保护柱 (八)峰展宽 1.进样体积过大 同(四)1 2.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散 3.数据系统采样速率太慢 设定速率应是每峰大于10点 4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10% 5.流动相粘度过高 增加柱温,采用低粘度流动相 6.检测池体积过大 用小体积池,卸下热交换器 7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱 8.柱外体积过大 将连接管径和连接管长度降至最小 9.样品过载 进小浓度小体积样品
(一)保留时间变化 1.柱温变化 柱恒温,必要时需配置恒温箱2.等度与梯度间未能充分平衡 至少用10倍柱体积的流动相平衡柱3.缓冲液容量不够 用25mmol/L的缓冲液4.柱污染 每天冲洗柱5.柱内条件变化 稳定进样条件,调节流动相6.柱快达到寿命 采用保护柱(二)保留时间缩短 1.流速增加 检查泵,重新设定流速2.样品超载 降低样品量3.键合相流失 流动相PH值保持在3~7.5检查柱的方向4.流动相组成变化 防止流动相蒸发或沉淀5.温度增加 柱恒温(三)保留时间延长 1.流速下降 管路泄漏,更换泵密封圈,排除泵内气泡2.硅胶柱上活性点变化 用流动相改性剂,如加三乙胺,或采用碱至钝化柱3.键合相流失 同前(二)34.流动相组成变化 同前(二)45.温度降低 同前(二)5(四) 出现肩峰或分叉 1.样品体积过大 用流动相配样,总的样品体积小于第一峰的15%2.样品溶剂过强 采用较弱的样品溶剂3.柱塌陷或形成短路通道 更换色谱柱,采用较弱腐蚀性条件4.柱内烧结不锈钢失效 更换烧结不锈钢,加在线过滤器,过滤样品5.进样器损坏 更换进样器转子(五)鬼峰1.进样阀残余峰 每次用后用强溶剂清洗阀,改进阀和样品的清洗2.样品中未知物 处理样品3.柱未平衡 重新平衡柱,用流动相作样品溶剂 (尤其是离子对色谱)4.三氟乙酸(TFA)氧化(肽谱) 每天新配,用抗氧化剂5.水污染(反相) 通过变化平衡时间检查水质量,用HPLC级的水(六) 基线噪声 1.气泡(尖锐峰) 流动相脱气,加柱后背压2.污染(随机噪声) 清洗柱,净化样品,用HPLC级试剂3.检测器灯连续噪声 更换氘灯4.电干扰(偶然噪声) 采用稳压电源,检查干扰的来源(如水浴等)5.检测器中有气泡 流动相脱气,加柱后背压(七)峰拖尾 1.柱超载 降低样品量,增加柱直径采用较高容量的固定相2.峰干扰 清洁样品,调整流动相3.硅羟基作用 加三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相PH值,钝化样品4.同前(四)4 同前(四)45.同前(四)3 5.同前(四)36.死体积或柱外体积过大 连接点降至最低,对所有连接点作合适调整,尽可能采用细内径的连接管7.柱效下降 用较低腐蚀条件,更换柱,采用保护柱(八)峰展宽 1.进样体积过大 同(四)12.在进样阀中造成峰扩展 进样前后排出气泡以降低扩散3.数据系统采样速率太慢 设定速率应是每峰大于10点4.检测器时间常数过大 设定时间常数为感兴趣第一峰半宽的10%5.流动相粘度过高 增加柱温,采用低粘度流动相6.检测池体积过大 用小体积池,卸下热交换器7.保留时间过长 等度洗脱时增加溶剂含量也可用梯度洗脱8.柱外体积过大 将连接管径和连接管长度降至最小9.样品过载 进小浓度小体积样品

盐雾腐蚀试验箱成果检查办法有四种类型:一、评级断定法,二、称重断定法,三、腐蚀物呈现断定法:四、腐蚀数据计算剖析法。评级断定法把腐蚀面积与总面积百分比的百分数按特定的办法划分成相应的等级,以其间的一个等级作为断定依据,它适合对平板样品进行测量,而称重断定法是对商品通过腐蚀实验的前后质量进行称重的办法,照着商品试验前后的分量对耐腐蚀性进行检定。[align=center][img=,348,348]https://ng1.17img.cn/bbsfiles/images/2021/02/202102261413253489_4681_1037_3.jpg!w348x348.jpg[/img][/align] 它对于金属的耐腐蚀功能检查十分适用,腐蚀物呈现断定法是定性断定法,在样品进行完盐雾腐蚀实验后,查看商品有没有腐蚀的状况来对商品进行断定,它首要应用于剖析和计算腐蚀状况,而不是特定的用于某一具体商品的质量而断定。 人工加快模仿盐雾环境试验的是使用具有必定的空间容积的设备就是盐雾腐蚀试验箱,人工制造出了盐雾环境对于样品的耐盐雾腐蚀性的断定。与天然环境进行对比,发现其氯化钠的盐浓度是通常天然环境盐雾试验的几倍乃至几十倍之后,加快了药品腐蚀的速度,对样品的耐盐雾腐蚀度的成果检查时刻也缩短了。在天然曝露环境下对样品进行耐腐蚀实验,等到其腐蚀或许要一年,但是人工模仿的话,一天之内就可得到相似的成果。 盐雾腐蚀试验箱性能指标: 1、盐水喷雾试验:NSS、ACSS。试验室:35℃±1℃。压力空气桶:47℃±1℃。 2、温度均匀度:≤±2℃ 3、温度波动度:≤±0.5℃ 4、盐雾的沉降量:1~2ml/80cm2.h 5、喷雾方式:气流喷雾,连续喷雾或者间歇喷雾。 6、试验定时:1~9999(S、M、H)可设可调。 7、耐腐蚀试验:CASS。试验室:50℃±1℃。压力空气桶:63℃±1℃。 我司专业从事环试设备生产和维修服务,我们真诚的希望广大客户可以信任我们!
EDX谱中有些元素的L峰甚至比K峰还更高更明显,而其K峰可能和另外元素的峰离得很近的情况下,能否断定这种元素的存在呢?例如Ti和N的K峰就离得很近,而又观察到Ti的L峰的情况下?怎么断定呢? 另K峰称为主峰?为什么?以前没怎么注意,觉得K峰一定要是最明显的,现在好像不是这样....另外,EDX对于一些元素,尤其是重元素,当其K峰的位置为几十Kev的时候,好像一般强度比较低吧,怎么断定这种元素是否存在?
各位大侠,在做样时出现一个峰正好与标准品出的峰处重合,所有的样品都是如此,且出的峰很高,这种现象能否断定这个峰就是要检测出的峰 如何断定它是杂质峰?
家居产品代理企业 “达芬奇”现象在我们的分析仪器里面有多少?我们遇见不少名为外企品牌 实为国产产品的仪器 垄断定价 在中国攫取超级利润。国内仪器代工企业不少,穿上外企马甲身价倍增,产品被垄断定价,利用国外强力推销渠道、国内代理 ,赚取超额利润。保税区里转一圈就身价倍增,我们能罗列出多少产品
比表面积测试仪有许多的方式供我们选用,通常我们选用的就是动态法、直接对比法、 多点BET法、静态容量法等多种方式,而今天我们所要学习的就是关于动态法的一些常见方式解决方案。 我们选用的动态法其实过程也不是那么复杂,只是需要我们更多的细心和解决方式。 比表面积测试仪首先就是将待测粉体样品装在U型的样品管内,使富含必定份额吸附质的混合气体流过样品,这样形成一种特地的测试效果,我们可以依据吸附前后气体浓度改变来断定被测样品对吸附质分子的吸附量来达到我们所要测试的成果。 比表面积测试仪静态法主要依据断定吸附吸附量办法的不一样分为分量法和容量法; 分量法是依据吸附前后样品分量改变来断定被测样品对吸附质分子的吸附量,来判断其测试的成分内容,更多的是因为分辨率低、准确度差、对设备需求很高级缺点已很少运用。所以很好的办法就是我们解决其弊端,然后达到我们所要用的要求,才能达到我们比表面的测试效果。 比表面积测试仪容量法是将待测粉体样品装在必定体积的一段关闭的试管状样品管内,然后通过向样品管内写入必定压力的吸附质气体,能给我们依据吸附前后的压力或分量改变来断定被测样品对吸附质分子的吸附量来达到我们所要进行的有效措施。 介绍了这么多关于比表面积测试仪的一些常见测试方法,更多的是要我们有效的改善我们的测试方式,达到我们更加仔细的能力,还有就是方面我们正常的工作和测试内容。www.chinazhongqi.net/93.html
如题,我的四极质谱仪出现了问题, 检测不出气体.你们知道如何断定灯丝烧断了呢?
不好断定pm10或PM2.5测试仪中的一台是否正常,更换取气通道后能用另一台测试验证嘛
做了两个样,保留时间分别是17.75min、17.34min,从UV上看,吸收峰波长相同,请问,能断定是同一物质吗?
日本政府敦促中方就问题饺子事件提供报告日本共同网报道,据多名日中外交消息人士31号透露,鉴于“问题饺子事件”发生近两年后真相依旧不明,日本外相冈田克也已经就问题饺子事件向中方提出要求,希望中方尽快完成中期报告,承认有问题物质是从中国混入。日方并希望在首相鸠山由纪夫明年访华前拿到报告。日本千叶县警察搜查1科去年5月15日宣布:在发生了中毒症状的千叶市稻毛区母子吃剩的冷冻饺子中,发现了相当于农药残存基准10万倍以上的有机磷系杀虫剂甲胺磷,是日本国内在这次中毒事件的药物分析中所获得的最高值。千叶县警方因此断定,甲胺磷是被人故意混入的。这是日本警察当局首次断定中国冷冻饺子中毒事件是一起人为的犯罪。
在原药材的挥发油的测定方法中,密度大于1和小于1的方法不一样,那我在做样品之前怎么去断定这个挥发油的密度呢?
最近我第一次采用薄层色谱进行物质定性定量分析样品,如果样品的斑点和标准样品的斑点距离相差很大,是否断定两边点不是同一物质?
版面有不少帖子讨论元素灯出毛病,那么,大家平时是如何判断元素灯出毛病,如何断定它不能用了呢?欢迎讨论!
看焦点访谈采访的时候里面出现了一个红外光谱仪,药厂的人不会用,也没有标准图谱。请问,有红外光谱仪和标准图谱就能直接断定二甘醇的存在吗?如果能 ,如何定量呢
我们用的是S-3000N扫描电镜,现在分析的时候为什么开到2小时左右电流就一直下降?一般换过灯丝之后电流在90uA左右但是一直降直到烧断为止,现在初步可以断定的是真空度可能有问题,但是具体的还要请各位详细说说?
做表面刻蚀的。好多文献里说关闭反馈?请问这个怎么关?把I和P都设为0么?关反馈意味着什么啊?另外问,STM扫描石墨原子的时候应该注意一些什么啊?我是怎么扫都扫不出来。大侠们都怎么来断定针尖是否状态好呢?
现在用的柱子是 60米 初步断定是柱效下降了。想换个新柱子在仪器备箱里找到一个同样材质的柱子 可就是长度是30mi 比现用的缩短一半大家说这个能用吗,还有如果用的话 要不要先老化一下柱子 再进样呢?



 我要推广仪器
我要推广仪器
 下载APP
下载APP